In my last notebook entry, I reviewed some basic initial analyses of Project Runway DNA sequencing data. One notable result was that the number of reads assigned to human-infecting viruses differed significantly between the pipeline I ran for that entry and the current public pipeline. In this entry, I dig into these differences in more depth, to see whether they tell us anything about which tools to incorporate into the next version of the public pipeline.
At a high level, there are three main differences between the two pipelines:
The public pipeline uses AdapterRemoval for removal of adapters and quality trimming, while my pipeline uses FASTP.
My pipeline uses bbduk to identify and remove ribosomal reads prior to Kraken analysis, while the public pipeline does not.
My pipeline applies deduplication prior to Kraken analysis using clumpify, while the public pipeline applies it after Kraken analysis via a manual method.
In principle, any of these differences could be responsible for the differences in read assignment we observe. However, since very few reads were identified as ribosomal or as duplicates by the new pipeline, it’s unlikely that these differences are those responsible.
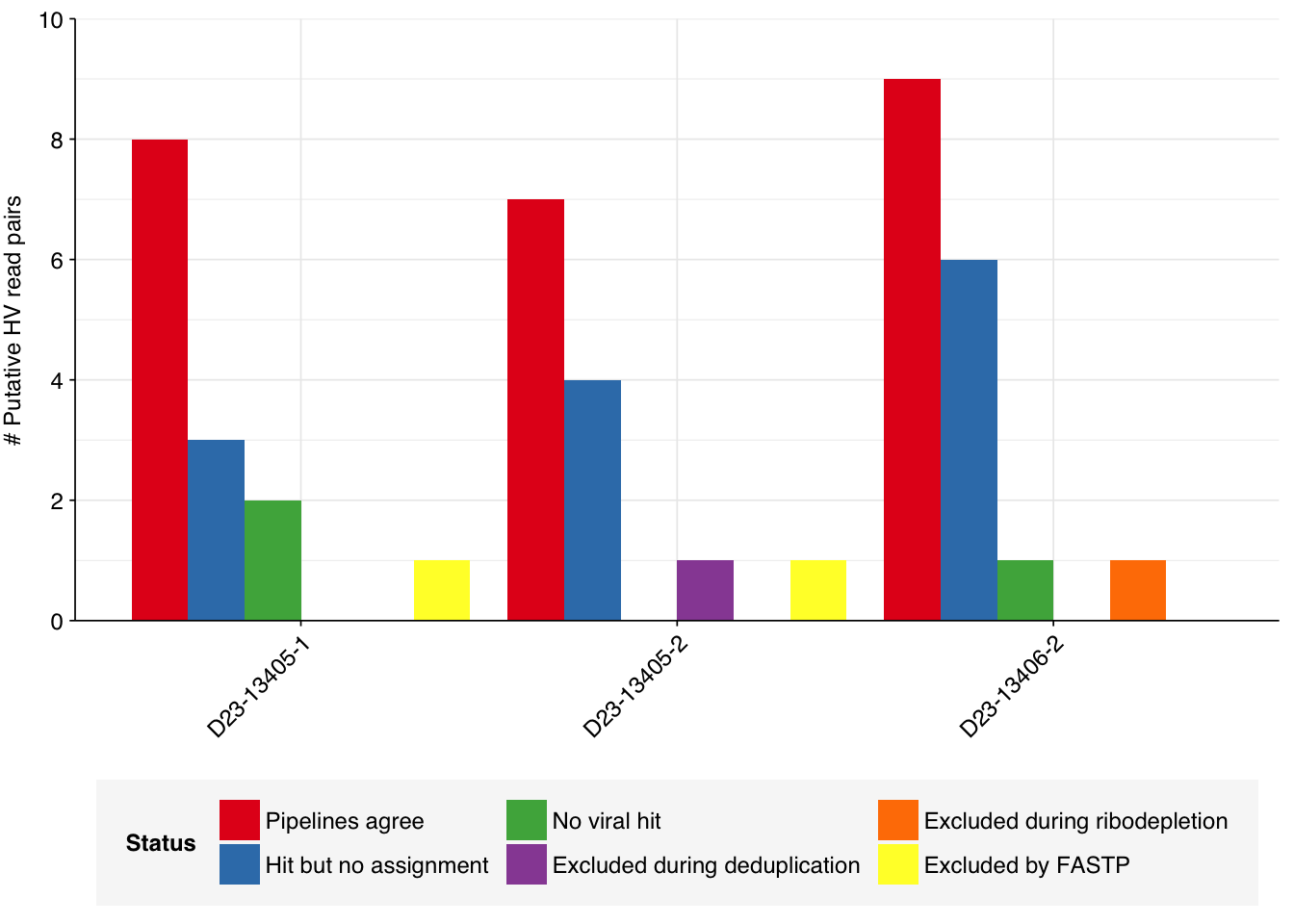
To investigate this, I decided to manually identify and trace the reads assigned to human-infecting viruses in both pipelines, to see whether that tells us anything about the likely cause of the differences. To do this, I selected the three samples from the dataset that show the largest difference in the number of assigned human-infecting virus reads (henceforth HV reads):
D23-13405-1 (14 HV reads assigned by the public pipeline vs 8 by the new pipeline)
D23-13405-2 (12 vs 8)
D23-13406-2 (17 vs 9)
The way the public pipeline does deduplication (after Kraken2 analysis, during dashboard generation) makes it difficult to directly extract the final list of HV read IDs for that pipeline, but it is quite easy to do this for the list of reads immediately prior to deduplication. Doing this for the samples specified above returned the following results:
D23-13405-1: 14 read IDs (no reads lost during deduplication)
D23-13405-2: 14 read IDs (2 reads to Simian Agent 10 lost during deduplication)
D23-13406-2: 17 read IDs (no reads lost during deduplication)
In the first and third of these cases, I could thus directly compare the Kraken output from the two pipelines to investigate the source of the disagreements. In the second case, it wasn’t immediately possible to identify which two out of the three Simian Agent 10 reads were considered duplicates in the dashboard, but I was at least able to restrict the possibility to those three reads. (As we’ll see below, information from the new pipeline also helped narrow this down.)
In this case, 8 HV reads were assigned by the new pipeline, and 14 reads were assigned by the the public pipeline.
All 8 of the reads assigned by the new pipeline were among the 14 HV reads assigned by the public pipeline.
Among the 6 remaining reads that were assigned by only the public pipeline:
3 appeared in the list of HV hits for the new pipeline; that is to say, for these four reads, the new pipeline was able to identify hits to human-infecting viruses but not make an overall assignment.
2 were included in the Kraken2 output for the new pipeline, but were not found to contain any HV k-mers and so were excluded from the list of hits. This is a more extreme case of the above situation: in this case, more stringent trimming by FASTQ has removed the putative HV k-mers.
1 was found among the reads that FASTP discarded due to not passing quality filters; in this case, read 2 was discarded due to low quality, and read 1 was then discarded due to lacking a read pair.
In all 6 cases, therefore, the difference in assignment between the two pipelines was found to be due to difference 1, i.e. the use of different preprocessing tools. In general, FASTP appears to be more stringent than AdapterRemoval in a way that resulted in fewer HV read assignments. But are these reads false positives for the old pipeline, or false negatives for the new one?
To address this, I extracted the raw sequences of the six read pairs, manually removed adapters, and manually analyzed them with NCBI BLAST (blastn vs viral nt, then vs full nt).
In all six cases, no match was found by blastn between the read sequence and the human-infecting virus putatively assigned by Kraken2 in the public pipeline. In four out of six cases, the best match for the read was to a bacterial sequence; in one case, the best match for the forward read was bacterial while the reverse matched a phage; and in one case no significant match was found for either read.
These results suggest to me that FASTP’s more stringent trimming and filtering is ensuring true negatives, rather than causing false ones.
D23-13405-2
In this case, 8 HV reads were assigned by the new pipeline, and 14 by the public pipeline excluding deduplication; 2 of the latter were removed during deduplication for the dashboard.
All 8 of the reads assigned by the new pipeline were among the 14 pre-deduplication HV reads assigned by the public pipeline; however, two of these were among the group of three reads (all to Simian Agent 10) that were collapsed into one by deduplication in the public pipeline.
This indicates that one of the reads present in the new pipeline results was removed by deduplication in the public pipeline results – that is to say, the two pipelines disagree slightly more than the raw HV read counts would suggest.
One read was removed as a duplicate by both pipelines; I discarded this one from consideration, bringing the number of read IDs for consideration down to 13.
Among the remaining 5 HV reads from the public pipeline:
4 appeared in the list of HV hits for the new pipeline; that is to say, for these four reads, the new pipeline was able to identify hits to human-infecting viruses but not make an overall assignment.
1 was discarded by FASTP during quality filtering: read 2 was discarded due to low quality, and read 1 was then discarded due to lacking a read pair.
As before, I extracted the raw reads corresponding to these 5 disagreements from the raw sequencing data, removed adapters manually, and ran the resulting sequences through NCBI BLAST (blastn vs viral nt, then vs full nt). 4 out of 5 read pairs showed no match to the assigned virus, while one showed a good, though partial, match.
In the case of the match, it looks like in both the public pipeline and the new pipeline, Kraken2 only identified a single k-mer from the origin virus (Sandfly fever Turkey virus); however, the public pipeline also identified 3 k-mers assigned to taxid 1 (root), while these were trimmed away in the new pipeline, and this was sufficient to prevent the overall assignment.
I’m not sure how to feel about this case. Ex ante, making an assignment on the basis of a single unique k-mer and three uninformative k-mers feels quite dubious, but ex post it does appear that at least part of the read was correctly assigned.
Investigating the pair of reads that were flagged as duplicates by the public pipeline but not the new pipeline, I found that they were a perfect match, but with the sequence of the forward read in one pair matching the reverse read in the second pair.
This was surprising to me, as it suggests that clumpify won’t remove duplicates if one is in reverse-complement to the other, which seems like a major oversight. I checked this, and indeed Clumpify fails to detect the duplicate in the current state but succeeds if I swap the forward and reverse reads for one of the read pairs.
This makes me less excited about using Clumpify for deduplication. UPDATE: I found an option for Clumpify that seems to solve this problem, at least in this case. See Conclusions for more.
It’s worth noting that, due to the way FASTQC analyses files, it will also fail to detect RC duplicates.
D23-13406-2
In this case, 9 HV reads were assigned by the new pipeline, and 17 by the public pipeline.
As before, I confirmed that all 9 HV reads assigned by the new pipeline were also assigned by the public pipeline, leaving 8 disagreements. Of these:
6 appeared in the list of reads for which the new pipeline found HV hits, but wasn’t able to make an overall assignment; in all six of these cases, the HV hits were to the taxon assigned by the public pipeline.
1 was included in the new pipeline’s Kraken output but had no viral hits.
The final clash is the most interesting, as this one was excluded not by FASTP or Kraken, but by bbduk: it was identified as ribosomal and discarded prior to deduplication.
As before, I extracted the raw reads corresponding to these 8 disagreements from the raw sequencing data, removed adapters manually, and ran the resulting sequences through NCBI BLAST (blastn vs viral nt, then vs nt).
For 7 of the 8 disagreements – all those arising from FASTP preprocessing – BLAST found no match between the read sequence and the taxon assigned by the public pipeline. As before, this suggests to be that FASTP is doing a good job preventing false positives through better preprocessing.
The BLAST result for the final disagreement is again the most interesting. The forward read indeed showed strong alignment to bacterial rRNA sequences, as found by bbduk. The reverse read, however, showed good alignment to Influenza A virus, which was the virus assigned by Kraken2 in the public pipeline. In this case, it appears we have a chimeric read, which both pipelines are in some sense processing correctly: bbduk is correctly identifying the forward read as ribosomal, and Kraken2 is correctly identifying the reverse read as viral. It’s not a priori obvious to me how we should handle these cases.
Sanity checking
Finally, I wanted to check that my findings here weren’t just the result of some issue with how I’m using NCBI BLAST – that is, that BLAST as I’m using it is able to detect true positives rather than just failing to find matches all over the place.
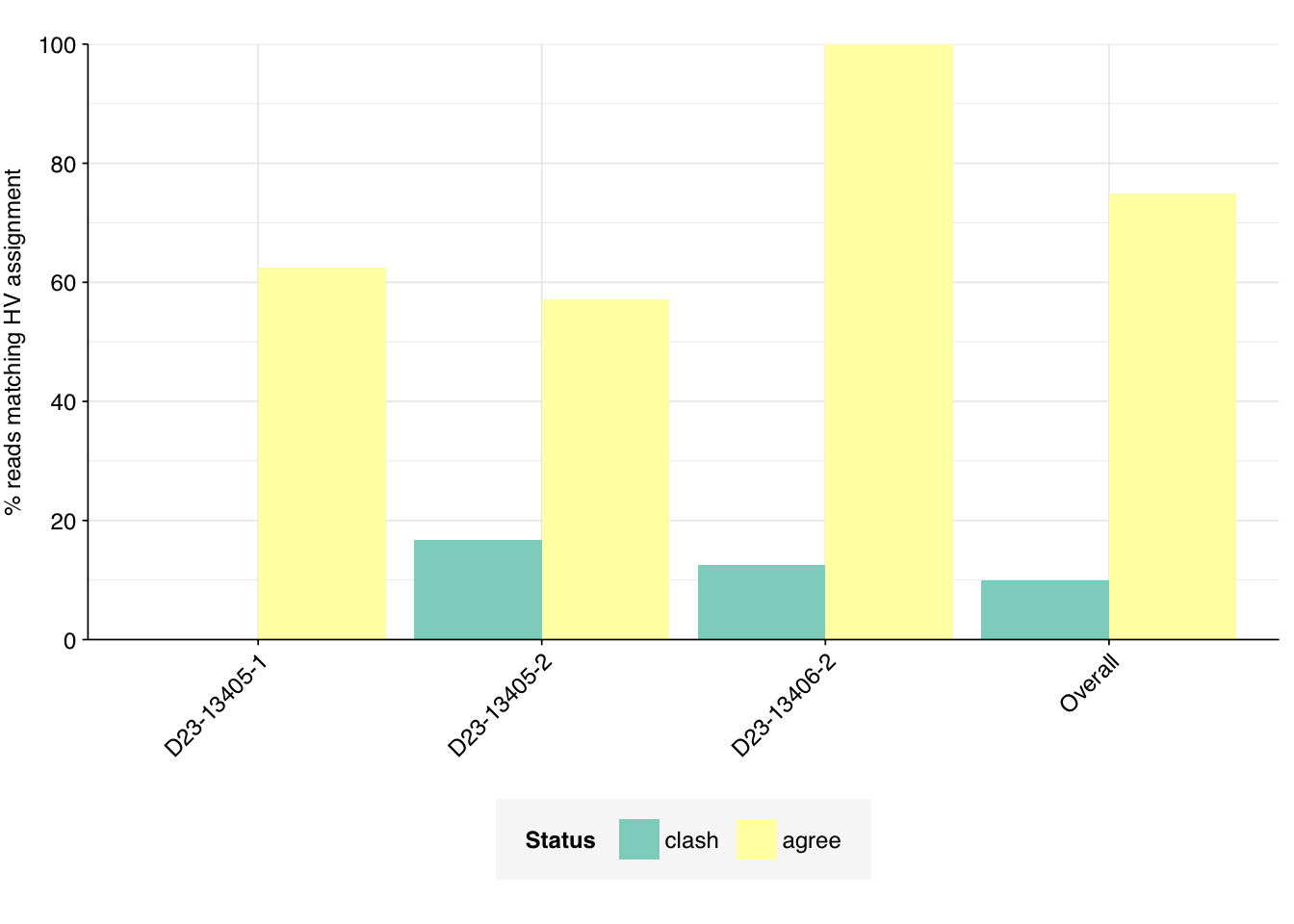
To check this, I took the 24 read pairs (8 + 7 + 9) from the three samples above that both pipelines agreed arose from human-infecting viruses, and ran these through BLAST in the same way as the disagreed-upon read-pairs above.
While the results weren’t as unequivocal as I’d hoped, they nevertheless showed a strong difference from those for the clashing read pairs. In total, 16/24 read pairs showed strong matching to the assigned virus, and an additional 2/24 showed a short partial match sufficient to explain the Kraken assignment. The remaining 6/24 failed to match the assigned virus. In total, 75% (18/24) of agreed-upon sequences showed a match to the assigned virus, compared to only 10% (2/20) for the clashing sequences.
This cautiously updates me further toward believing that the FASTP component of the new pipeline is mostly doing well at correctly rejecting true negatives, at least compared to the current public pipeline. That said, it also suggests that either (a) BLAST is generating a significant number of false negatives, or (b) even the new pipeline is generating a significant number of false positives.
My updates from this exercise are different for different parts of the pipeline.
For difference 1 (preprocessing tool) I mostly found that, in cases where the new pipeline rejects a read due to preprocessing and the public pipeline does not, that read appears to be a true negative. I’m not super confident about this, since I don’t 100% trust BLAST to not be producing false negatives here, but overall I think the evidence points to FASTP doing a better job here than AdapterRemoval.
I’d ideally like to shift to a version of the pipeline where we’re not relying on Kraken to make assignment decisions, and are instead running all Kraken hits through an alignment-based validation pipeline to determine final assignments. I’d be interested in seeing how these results look after making that change.
For difference 2 (ribodepletion) results here are equivocal. The single read pair I inspected that got removed during ribodepletion appears to include both a true ribosomal read and a true viral read. I think some internal discussion is needed to decide how to handle these cases.
For difference 3 (deduplication) I initially updated negatively about Clumpify, which appeared to be unable to handle duplicates where the forward and reverse reads in a pair are switched (this is also a case where FASTQC will be unable to detect these duplicates).
However, I found an option for Clumpify which addresses this issue, at least in this case. Specifically, one can configure Clumpify to unpair reads, perform deduplication on the forward and reverse reads all together, and then restore pairing. This successfully removes this class of duplicates.
I’m a little worried that this approach might sometimes cause complete loss of all duplicate reads (rather than all-but-one-pair) when the best-quality duplicate differs between forward and reverse reads. I tried this out by artificially modifying the quality scores for the duplicate reads from D23-13405-2, and this doesn’t seem to be the case at least in this instance: when quality across read pairs was concordant, I was able to control which read pair survived as expected, and when it was discordant, one read pair survived anyway. Still, this remains a niggling doubt.
Source Code
---title: "Comparing viral read assignments between pipelines on Project Runway data"subtitle: ""author: "Will Bradshaw"date: 2023-11-02format: html: code-fold: true code-tools: true code-link: true df-print: pagededitor: visualtitle-block-banner: black---```{r}#| label: load-packages#| include: falselibrary(tidyverse)library(cowplot)library(patchwork)library(fastqcr)source("../scripts/aux_plot-theme.R")```In my [last notebook entry](https://data.securebio.org/wills-public-notebook/notebooks/2023-10-24_project-runway-initial.html), I reviewed some basic initial analyses of Project Runway DNA sequencing data. One notable result was that the number of reads assigned to human-infecting viruses differed significantly between the pipeline I ran for that entry and the current [public pipeline](https://github.com/naobservatory/mgs-pipeline/). In this entry, I dig into these differences in more depth, to see whether they tell us anything about which tools to incorporate into the next version of the public pipeline.At a high level, there are three main differences between the two pipelines:1. The public pipeline uses AdapterRemoval for removal of adapters and quality trimming, while my pipeline uses FASTP.2. My pipeline uses bbduk to identify and remove ribosomal reads prior to Kraken analysis, while the public pipeline does not.3. My pipeline applies deduplication prior to Kraken analysis using clumpify, while the public pipeline applies it after Kraken analysis via a manual method.In principle, any of these differences could be responsible for the differences in read assignment we observe. However, since very few reads were identified as ribosomal or as duplicates by the new pipeline, it's unlikely that these differences are those responsible.To investigate this, I decided to manually identify and trace the reads assigned to human-infecting viruses in both pipelines, to see whether that tells us anything about the likely cause of the differences. To do this, I selected the three samples from the dataset that show the largest difference in the number of assigned human-infecting virus reads (henceforth HV reads):1. D23-13405-1 (14 HV reads assigned by the public pipeline vs 8 by the new pipeline)2. D23-13405-2 (12 vs 8)3. D23-13406-2 (17 vs 9)The way the public pipeline does deduplication (after Kraken2 analysis, during dashboard generation) makes it difficult to directly extract the final list of HV read IDs for that pipeline, but it is quite easy to do this for the list of reads immediately prior to deduplication. Doing this for the samples specified above returned the following results:1. D23-13405-1: 14 read IDs (no reads lost during deduplication)2. D23-13405-2: 14 read IDs (2 reads to Simian Agent 10 lost during deduplication)3. D23-13406-2: 17 read IDs (no reads lost during deduplication)In the first and third of these cases, I could thus directly compare the Kraken output from the two pipelines to investigate the source of the disagreements. In the second case, it wasn't immediately possible to identify which two out of the three Simian Agent 10 reads were considered duplicates in the dashboard, but I was at least able to restrict the possibility to those three reads. (As we'll see below, information from the new pipeline also helped narrow this down.)```{r}data_dir <-"../data/2023-11-01_pr-comp"read_status_path <-file.path(data_dir, "read-status.csv")read_status <-read_csv(read_status_path, show_col_types =FALSE) %>%mutate(status =fct_inorder(status))theme_kit <- theme_base +theme(aspect.ratio =1/2,axis.text.x =element_text(hjust =1, angle =45),axis.title.x =element_blank())g_status <-ggplot(read_status, aes(x=sample, y=n_reads, fill=status)) +geom_col(position ="dodge") +scale_fill_brewer(palette ="Set1", name ="Status") +scale_y_continuous(name ="# Putative HV read pairs", limits =c(0,10),breaks =seq(0,10,2), expand =c(0,0)) + theme_base + theme_kitg_status```# D23-13405-1- In this case, 8 HV reads were assigned by the new pipeline, and 14 reads were assigned by the the public pipeline.- All 8 of the reads assigned by the new pipeline were among the 14 HV reads assigned by the public pipeline.- Among the 6 remaining reads that were assigned by only the public pipeline: - 3 appeared in the list of HV hits for the new pipeline; that is to say, for these four reads, the new pipeline was able to identify hits to human-infecting viruses but not make an overall assignment. - 2 were included in the Kraken2 output for the new pipeline, but were not found to contain any HV k-mers and so were excluded from the list of hits. This is a more extreme case of the above situation: in this case, more stringent trimming by FASTQ has removed the putative HV k-mers. - 1 was found among the reads that FASTP discarded due to not passing quality filters; in this case, read 2 was discarded due to low quality, and read 1 was then discarded due to lacking a read pair.- In all 6 cases, therefore, the difference in assignment between the two pipelines was found to be due to difference 1, i.e. the use of different preprocessing tools. In general, FASTP appears to be more stringent than AdapterRemoval in a way that resulted in fewer HV read assignments. But are these reads false positives for the old pipeline, or false negatives for the new one? - To address this, I extracted the raw sequences of the six read pairs, manually removed adapters, and manually analyzed them with NCBI BLAST (blastn vs viral nt, then vs full nt). - In all six cases, no match was found by blastn between the read sequence and the human-infecting virus putatively assigned by Kraken2 in the public pipeline. In four out of six cases, the best match for the read was to a bacterial sequence; in one case, the best match for the forward read was bacterial while the reverse matched a phage; and in one case no significant match was found for either read. - These results suggest to me that FASTP's more stringent trimming and filtering is ensuring true negatives, rather than causing false ones.# D23-13405-2- In this case, 8 HV reads were assigned by the new pipeline, and 14 by the public pipeline excluding deduplication; 2 of the latter were removed during deduplication for the dashboard.- All 8 of the reads assigned by the new pipeline were among the 14 pre-deduplication HV reads assigned by the public pipeline; however, two of these were among the group of three reads (all to Simian Agent 10) that were collapsed into one by deduplication in the public pipeline. - This indicates that one of the reads present in the new pipeline results was removed by deduplication in the public pipeline results -- that is to say, the two pipelines disagree slightly more than the raw HV read counts would suggest. - One read was removed as a duplicate by both pipelines; I discarded this one from consideration, bringing the number of read IDs for consideration down to 13.- Among the remaining 5 HV reads from the public pipeline: - 4 appeared in the list of HV hits for the new pipeline; that is to say, for these four reads, the new pipeline was able to identify hits to human-infecting viruses but not make an overall assignment. - 1 was discarded by FASTP during quality filtering: read 2 was discarded due to low quality, and read 1 was then discarded due to lacking a read pair.- As before, I extracted the raw reads corresponding to these 5 disagreements from the raw sequencing data, removed adapters manually, and ran the resulting sequences through NCBI BLAST (blastn vs viral nt, then vs full nt). 4 out of 5 read pairs showed no match to the assigned virus, while one showed a good, though partial, match. - In the case of the match, it looks like in both the public pipeline and the new pipeline, Kraken2 only identified a single k-mer from the origin virus (Sandfly fever Turkey virus); however, the public pipeline also identified 3 k-mers assigned to taxid 1 (root), while these were trimmed away in the new pipeline, and this was sufficient to prevent the overall assignment. - I'm not sure how to feel about this case. Ex ante, making an assignment on the basis of a single unique k-mer and three uninformative k-mers feels quite dubious, but ex post it does appear that at least part of the read was correctly assigned.- Investigating the pair of reads that were flagged as duplicates by the public pipeline but not the new pipeline, I found that they were a perfect match, but with the sequence of the forward read in one pair matching the reverse read in the second pair. - This was surprising to me, as it suggests that clumpify won't remove duplicates if one is in reverse-complement to the other, which seems like a major oversight. I checked this, and indeed Clumpify fails to detect the duplicate in the current state but succeeds if I swap the forward and reverse reads for one of the read pairs. - This makes me less excited about using Clumpify for deduplication. UPDATE: I found an option for Clumpify that seems to solve this problem, at least in this case. See Conclusions for more. - It's worth noting that, due to the way FASTQC analyses files, it will also fail to detect RC duplicates.# D23-13406-2- In this case, 9 HV reads were assigned by the new pipeline, and 17 by the public pipeline.- As before, I confirmed that all 9 HV reads assigned by the new pipeline were also assigned by the public pipeline, leaving 8 disagreements. Of these: - 6 appeared in the list of reads for which the new pipeline found HV hits, but wasn't able to make an overall assignment; in all six of these cases, the HV hits were to the taxon assigned by the public pipeline. - 1 was included in the new pipeline's Kraken output but had no viral hits. - The final clash is the most interesting, as this one was excluded not by FASTP or Kraken, but by bbduk: it was identified as ribosomal and discarded prior to deduplication.- As before, I extracted the raw reads corresponding to these 8 disagreements from the raw sequencing data, removed adapters manually, and ran the resulting sequences through NCBI BLAST (blastn vs viral nt, then vs nt). - For 7 of the 8 disagreements -- all those arising from FASTP preprocessing -- BLAST found no match between the read sequence and the taxon assigned by the public pipeline. As before, this suggests to be that FASTP is doing a good job preventing false positives through better preprocessing. - The BLAST result for the final disagreement is again the most interesting. The forward read indeed showed strong alignment to bacterial rRNA sequences, as found by bbduk. The reverse read, however, showed good alignment to Influenza A virus, which was the virus assigned by Kraken2 in the public pipeline. In this case, it appears we have a chimeric read, which both pipelines are in some sense processing correctly: bbduk is correctly identifying the forward read as ribosomal, and Kraken2 is correctly identifying the reverse read as viral. It's not *a priori* obvious to me how we should handle these cases.# Sanity checking- Finally, I wanted to check that my findings here weren't just the result of some issue with how I'm using NCBI BLAST -- that is, that BLAST as I'm using it is able to detect true positives rather than just failing to find matches all over the place.- To check this, I took the 24 read pairs (8 + 7 + 9) from the three samples above that both pipelines agreed arose from human-infecting viruses, and ran these through BLAST in the same way as the disagreed-upon read-pairs above.- While the results weren't as unequivocal as I'd hoped, they nevertheless showed a strong difference from those for the clashing read pairs. In total, 16/24 read pairs showed strong matching to the assigned virus, and an additional 2/24 showed a short partial match sufficient to explain the Kraken assignment. The remaining 6/24 failed to match the assigned virus. In total, 75% (18/24) of agreed-upon sequences showed a match to the assigned virus, compared to only 10% (2/20) for the clashing sequences.- This cautiously updates me further toward believing that the FASTP component of the new pipeline is mostly doing well at correctly rejecting true negatives, at least compared to the current public pipeline. That said, it also suggests that either (a) BLAST is generating a significant number of false negatives, or (b) even the new pipeline is generating a significant number of false positives.```{r}read_hit_path <-file.path(data_dir, "read-hit-count.csv")read_hit <-read_csv(read_hit_path, show_col_types =FALSE) %>%mutate(status =fct_inorder(status),p_hit = n_hit/n_reads)g_hit <-ggplot(read_hit, aes(x=sample, y=p_hit, fill=status)) +geom_col(position ="dodge") +scale_fill_brewer(palette ="Set3", name ="Status") +scale_y_continuous(name ="% reads matching HV assignment", limits =c(0,1),breaks =seq(0,1,0.2), expand =c(0,0), labels =function(y) y*100) + theme_base + theme_kitg_hit```# Conclusions- My updates from this exercise are different for different parts of the pipeline.- For difference 1 (preprocessing tool) I mostly found that, in cases where the new pipeline rejects a read due to preprocessing and the public pipeline does not, that read appears to be a true negative. I'm not super confident about this, since I don't 100% trust BLAST to not be producing false negatives here, but overall I think the evidence points to FASTP doing a better job here than AdapterRemoval. - I'd ideally like to shift to a version of the pipeline where we're not relying on Kraken to make assignment decisions, and are instead running all Kraken hits through an alignment-based validation pipeline to determine final assignments. I'd be interested in seeing how these results look after making that change.- For difference 2 (ribodepletion) results here are equivocal. The single read pair I inspected that got removed during ribodepletion appears to include both a true ribosomal read and a true viral read. I think some internal discussion is needed to decide how to handle these cases.- For difference 3 (deduplication) I initially updated negatively about Clumpify, which appeared to be unable to handle duplicates where the forward and reverse reads in a pair are switched (this is also a case where FASTQC will be unable to detect these duplicates). - However, I found an option for Clumpify which addresses this issue, at least in this case. Specifically, one can configure Clumpify to unpair reads, perform deduplication on the forward and reverse reads all together, and then restore pairing. This successfully removes this class of duplicates. - I'm a little worried that this approach might sometimes cause complete loss of all duplicate reads (rather than all-but-one-pair) when the best-quality duplicate differs between forward and reverse reads. I tried this out by artificially modifying the quality scores for the duplicate reads from D23-13405-2, and this doesn't seem to be the case at least in this instance: when quality across read pairs was concordant, I was able to control which read pair survived as expected, and when it was discordant, one read pair survived anyway. Still, this remains a niggling doubt.