Continuing our look at air sampling datasets, we turn to Rosario et al. (2018), another study of air filters, this time from HVAC filters from an undergraduate dorm building at the University of Colorado campus in Boulder. As in Prussin, samples were eluted from filters (in this case MERV-8, so less stringent than Prussin’s MERV-14 filters) and underwent both RNA and DNA sequencing – this time on an Illumina MiSeq with 2x250bp reads.
The raw data
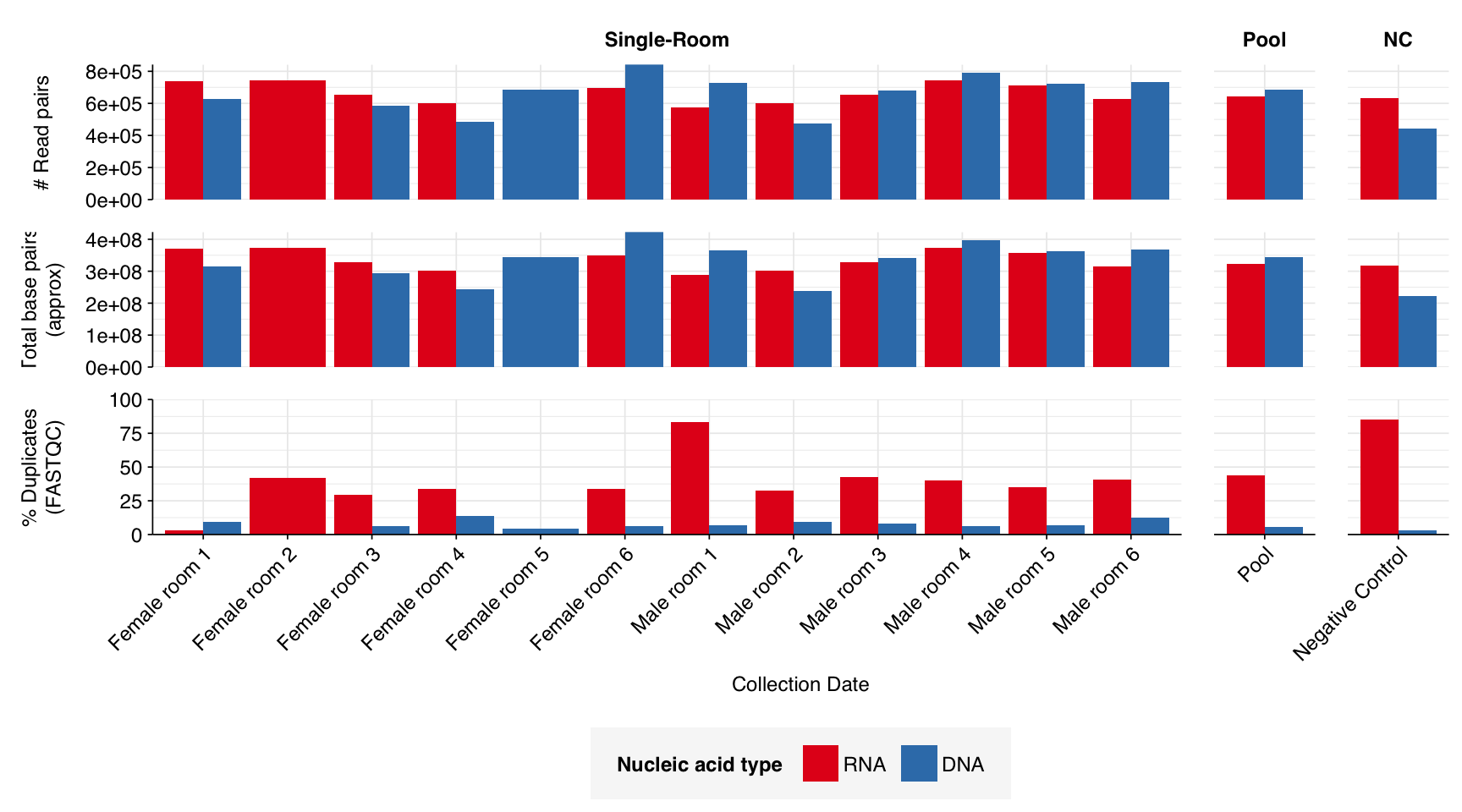
The Rosario dataset comprised sequencing data from 12 individual dormitory rooms at the sampling site, as well as a pooled sample of eight additional rooms and a negative control.
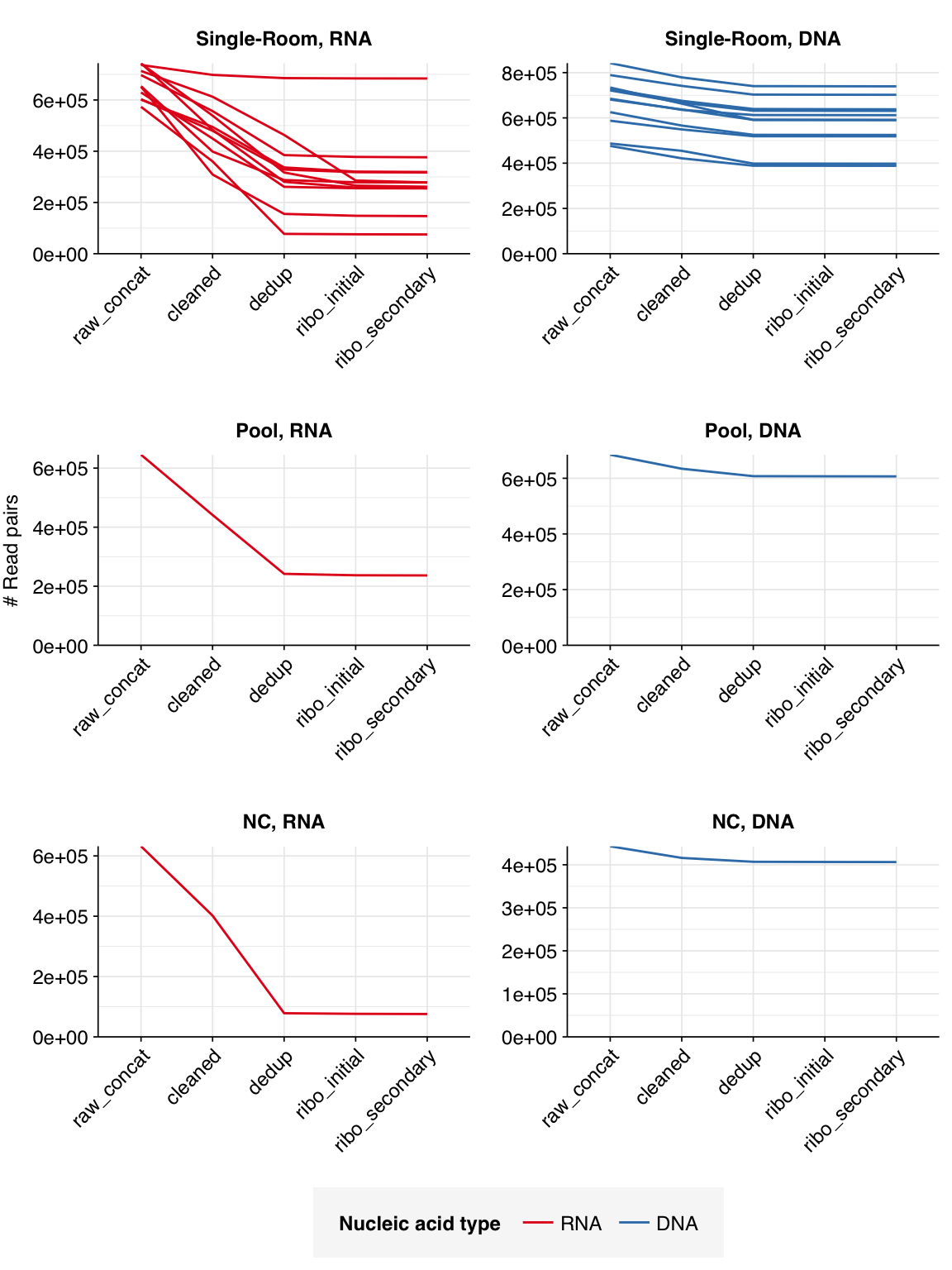
The 12 single-room samples yielded 0.57-0.74M (mean 0.67M) RNA-sequencing reads and 0.48M-0.84M (mean 0.67M) DNA-sequencing reads; the pooled and control samples had similar depth. In total, non-control samples yielded 8.0M RNA read pairs and 8.0M DNA read pairs (4 gigabases of sequence each).
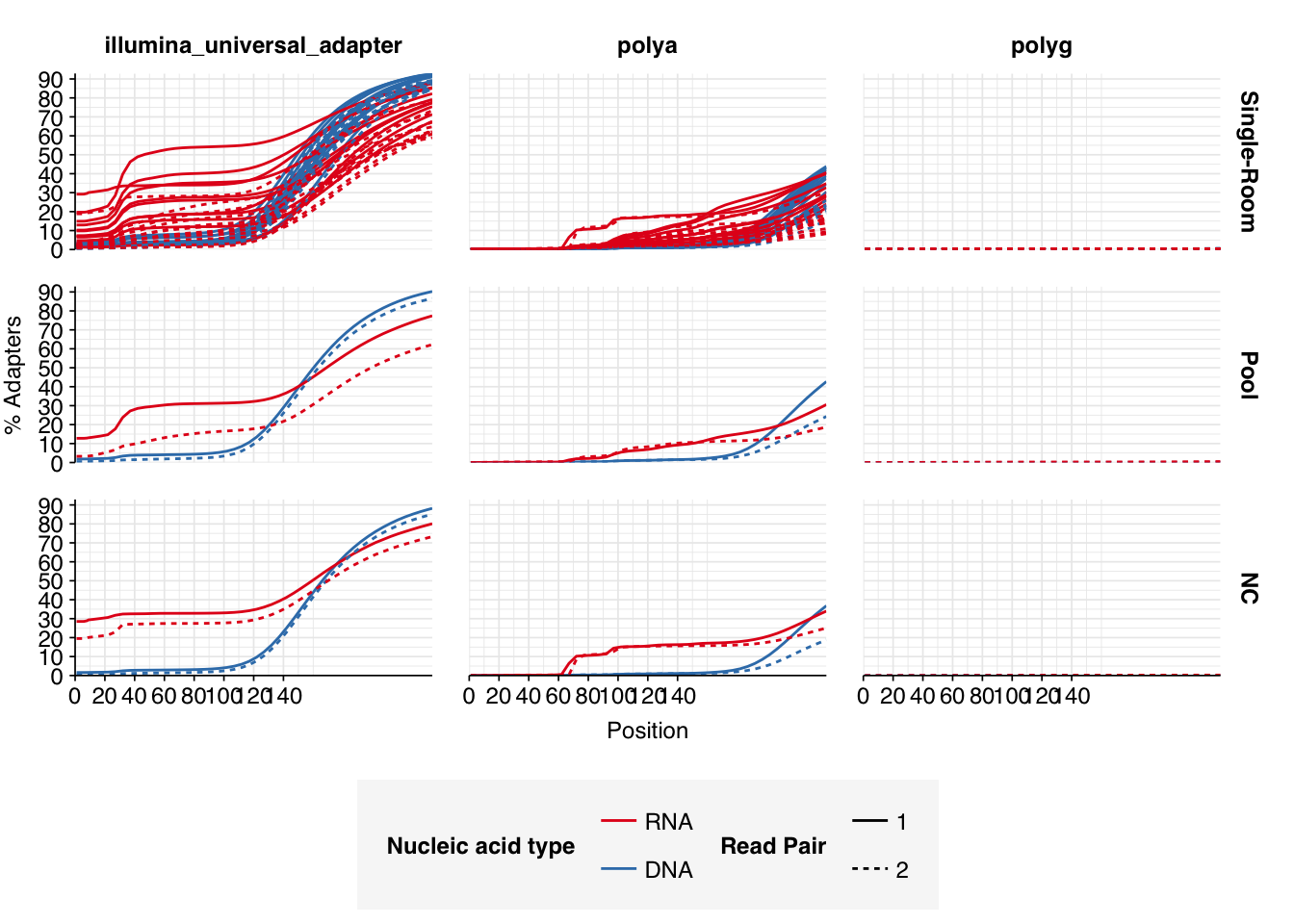
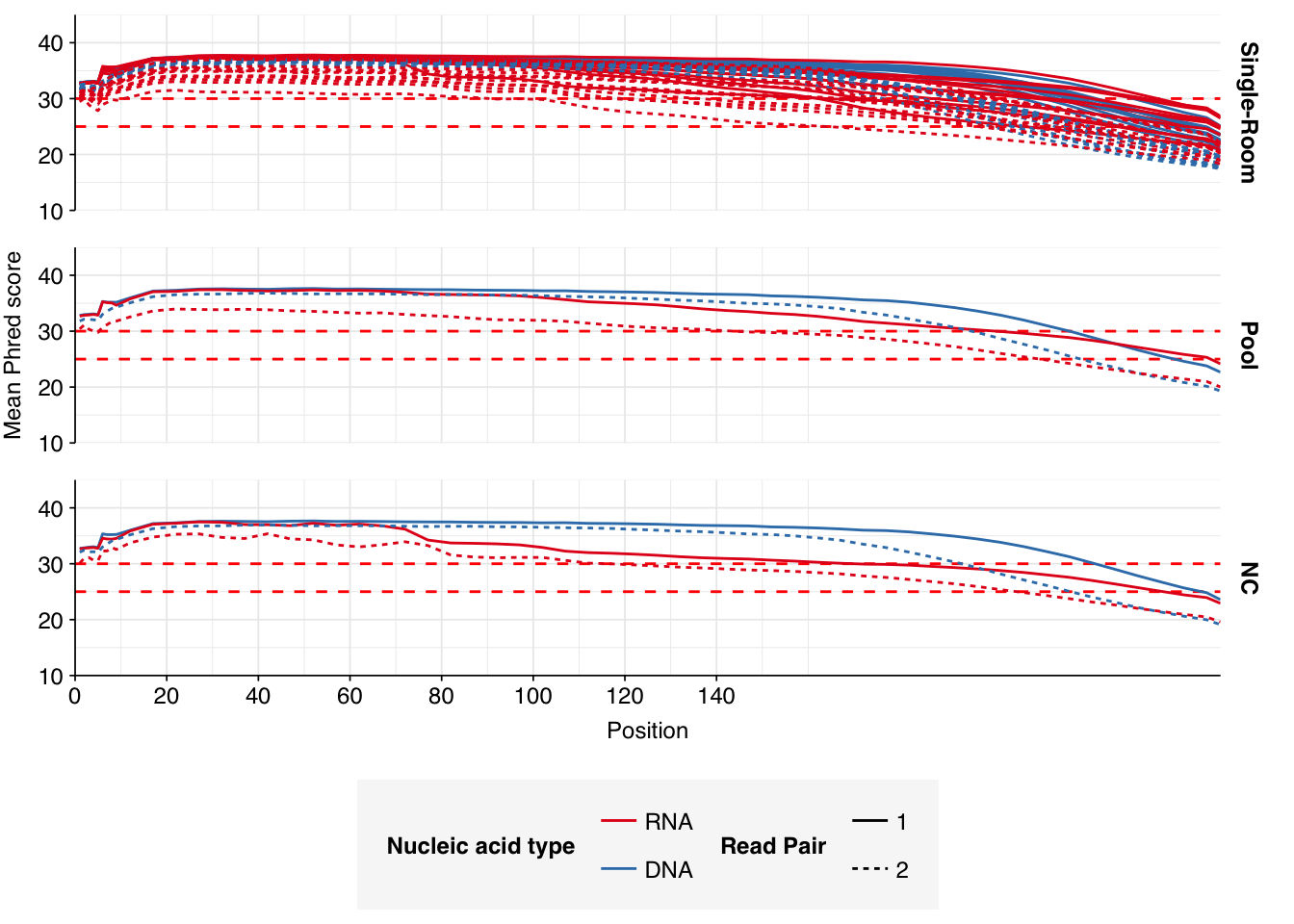
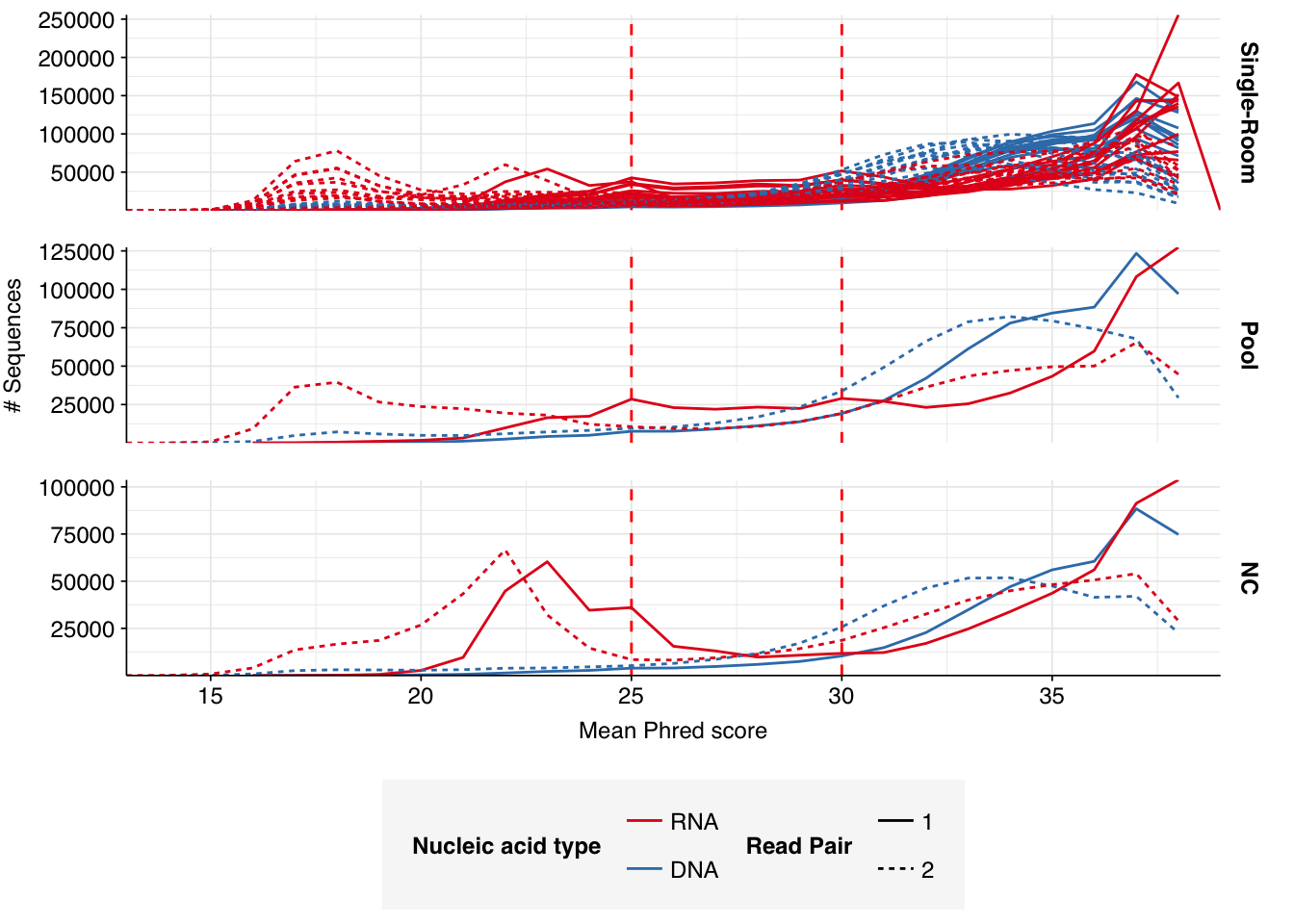
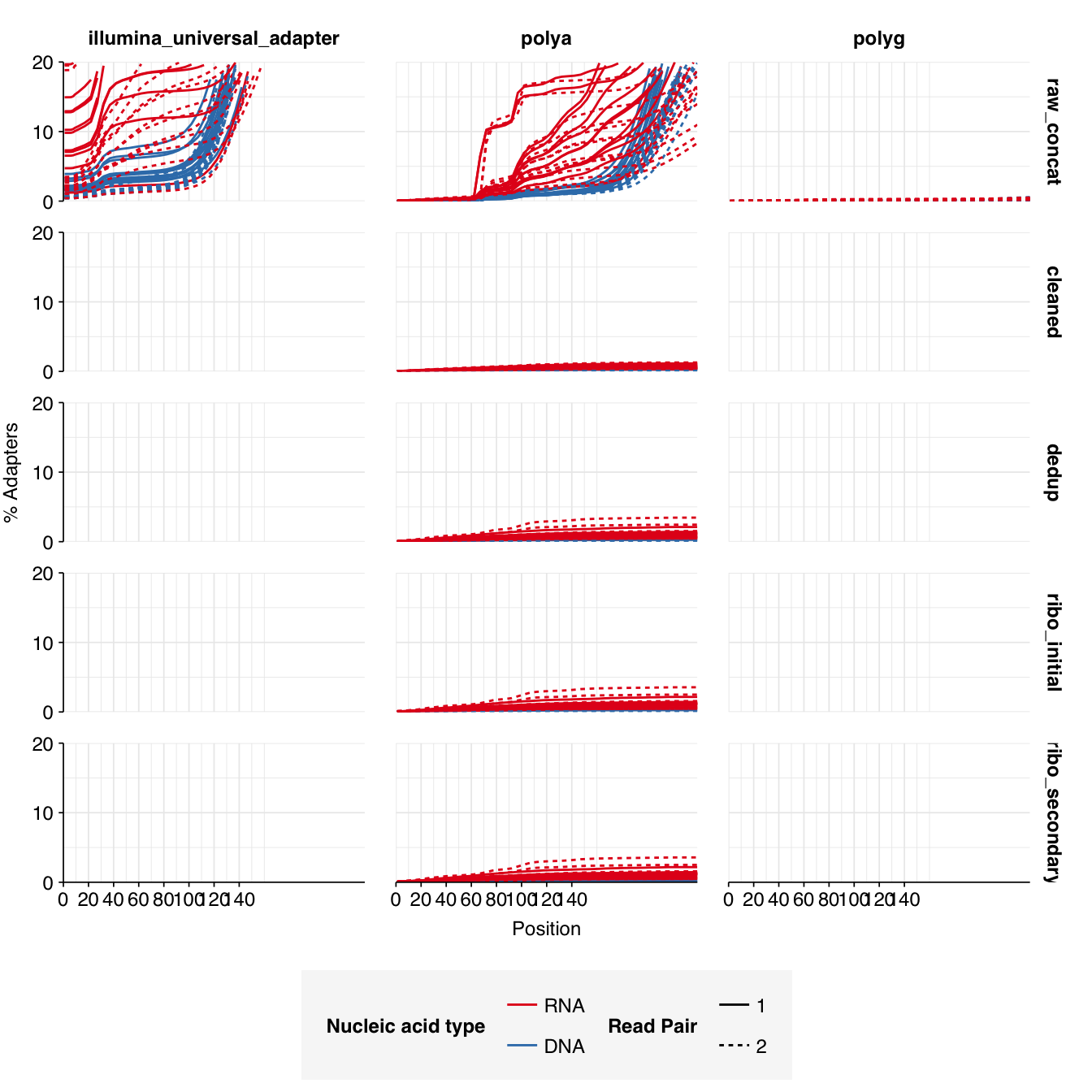
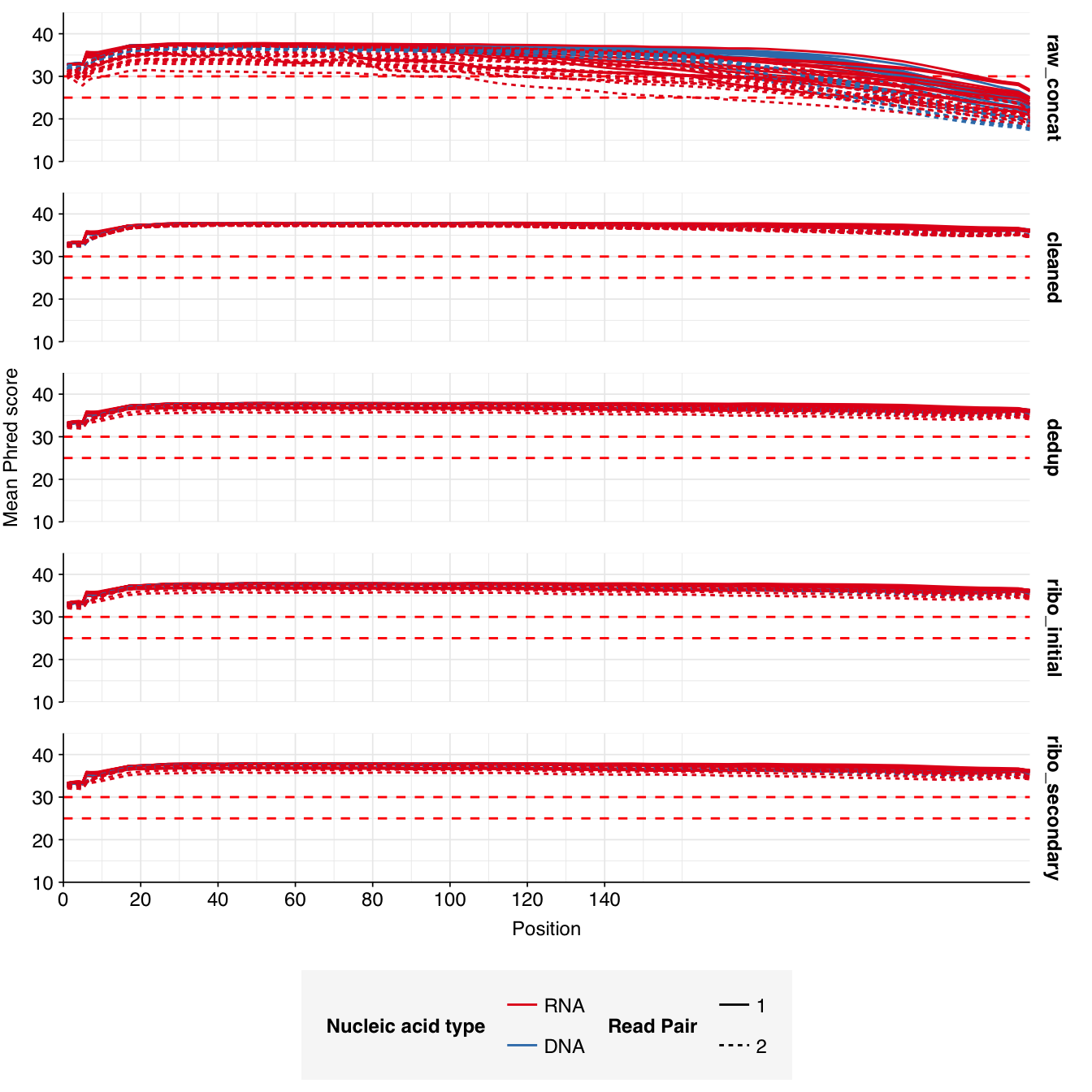
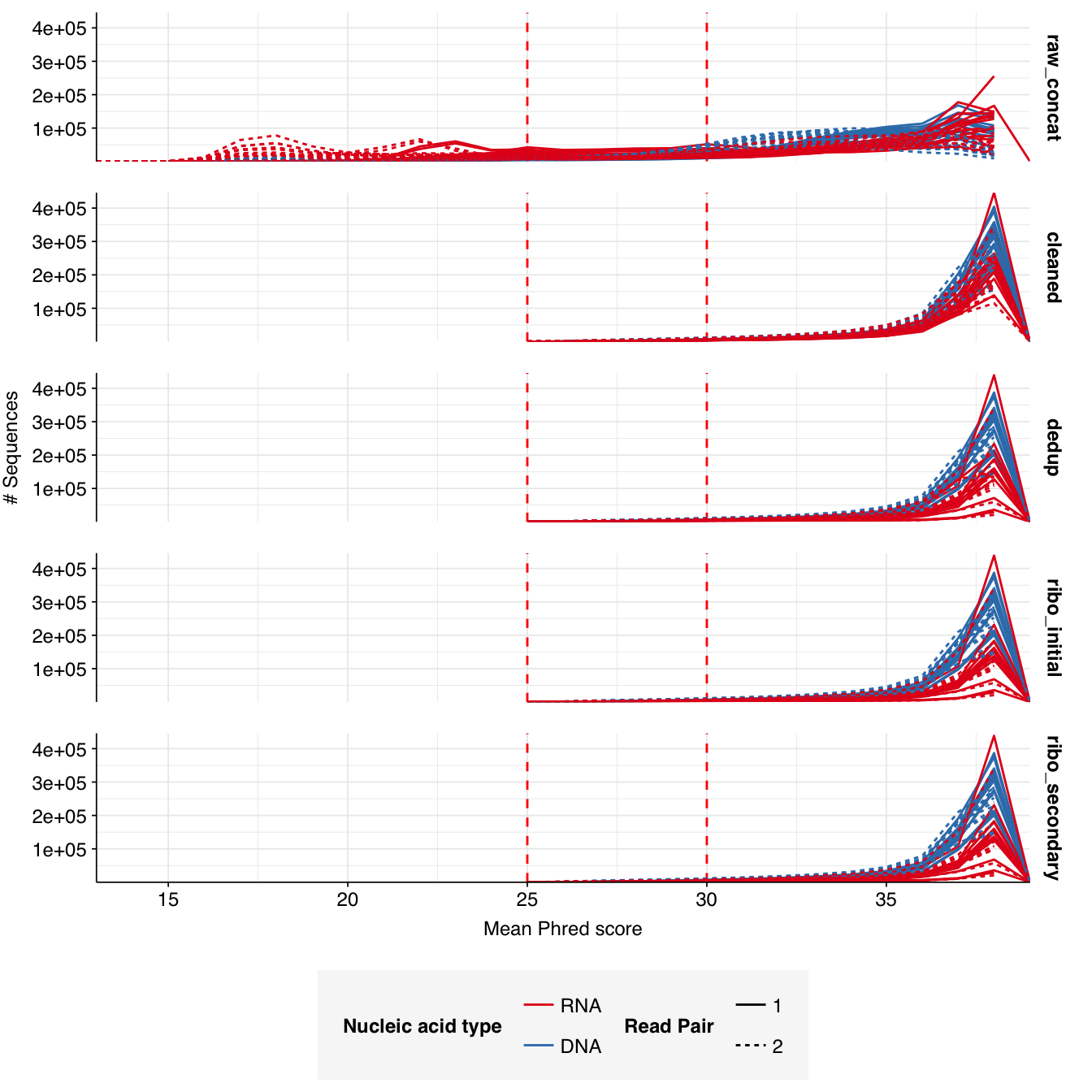
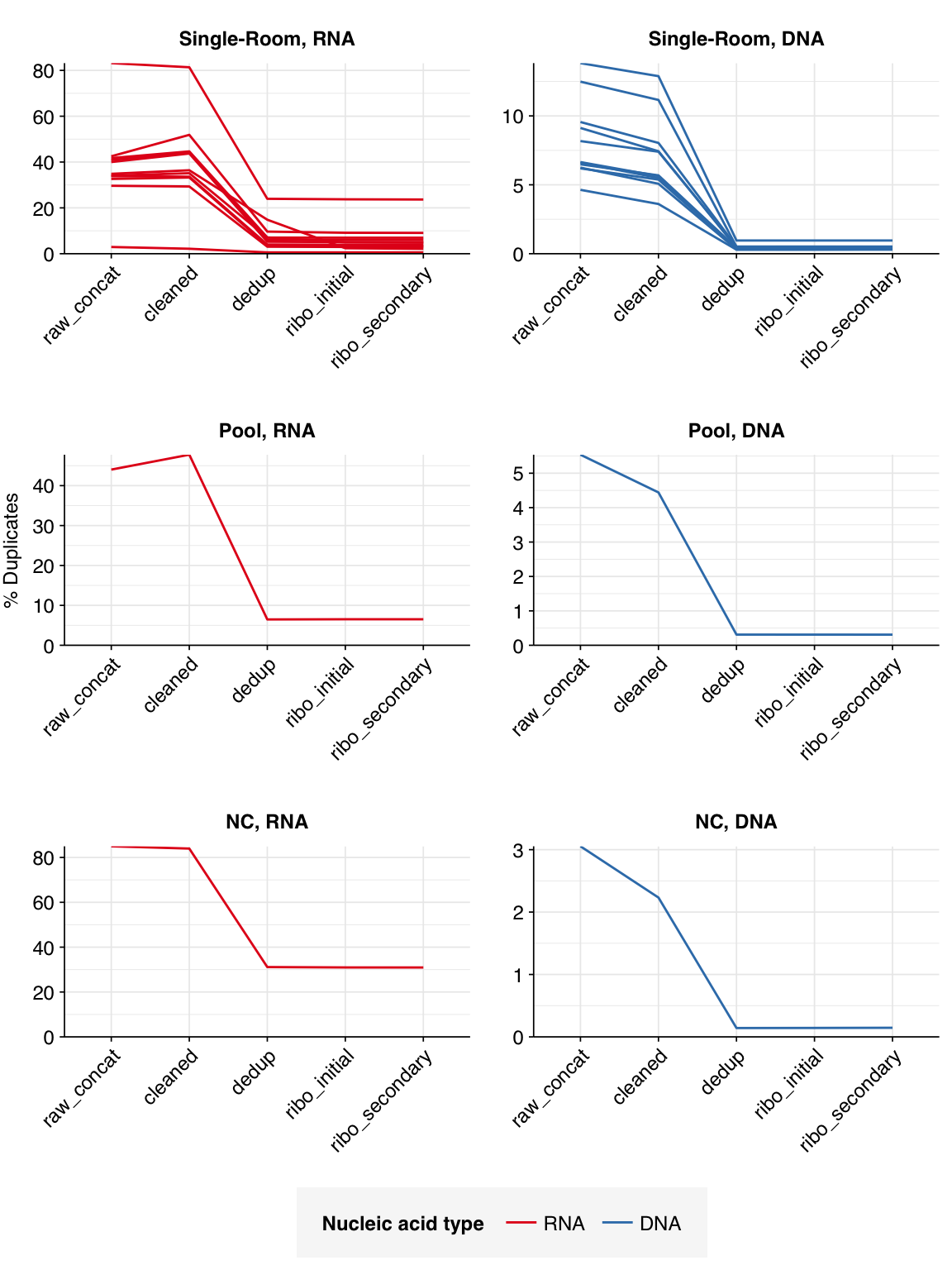
Read qualities were so-so, in need of cleaning. Adapter levels were high. Inferred duplication levels were low (4.6-8.2%) in DNA reads but highly variable (3-83%) in RNA reads.
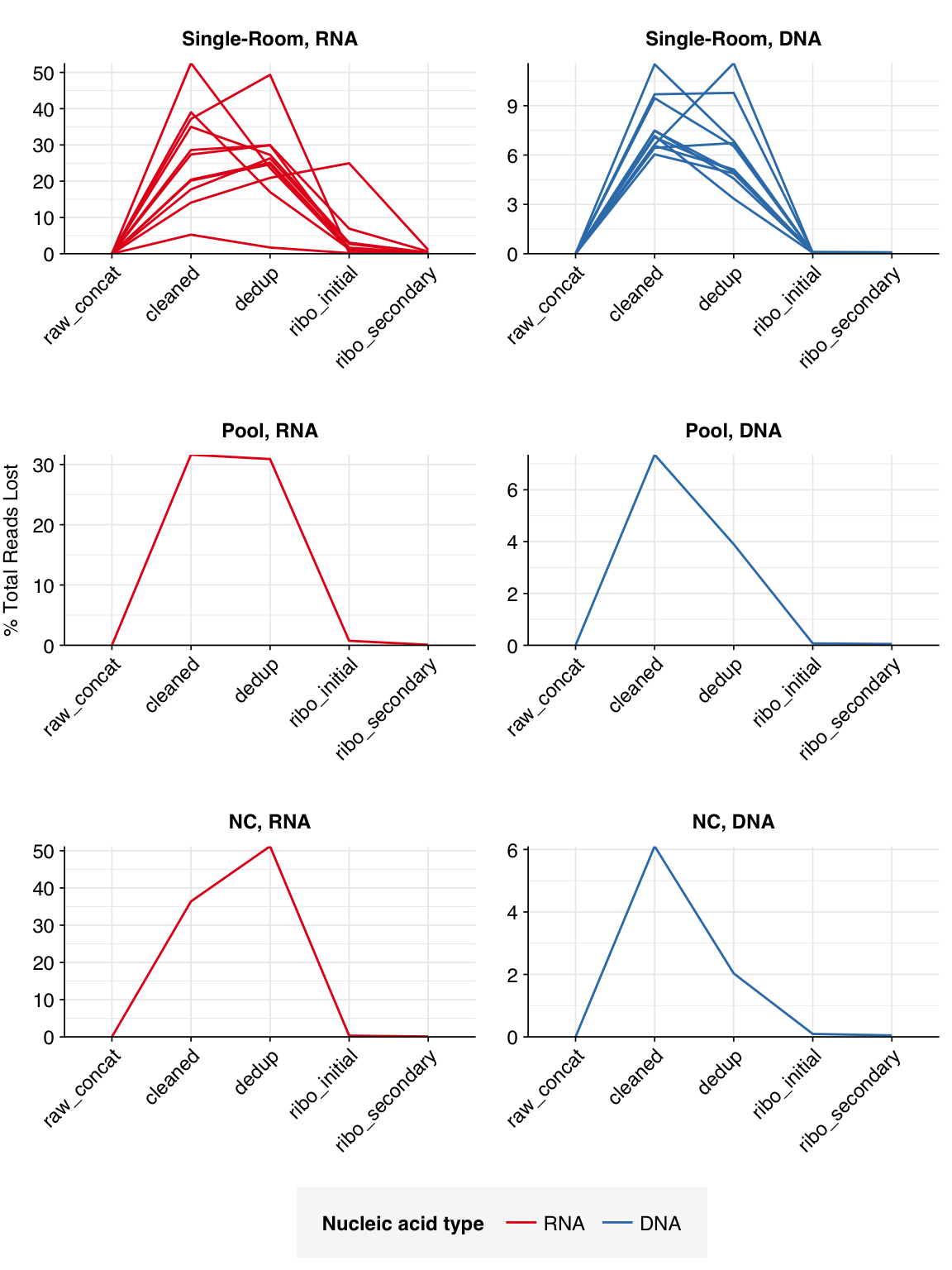
The average fraction of reads lost at each stage in the preprocessing pipeline is shown in the following table. Single-room samples lost an average of 52%/14% (RNA/DNA) of total read pairs during cleaning and deduplication, with a further loss of 4.3%/0.1% during ribodepletion. However, the fraction of reads lost, especially during cleaning and deduplication, was highly variable between samples.
# Plot relative read losses during preprocessingg_reads_rel<-ggplot(n_reads_rel, aes(x=stage, color=na_type, group=sample))+geom_line(aes(y=p_reads_lost_abs_marginal))+scale_y_continuous("% Total Reads Lost", expand=c(0,0), labels =function(x)x*100)+scale_color_na()+facet_wrap(sample_type~na_type, scales="free", ncol=2, labeller =label_wrap_gen(multi_line=FALSE))+theme_kitg_reads_rel
Data cleaning was very successful at improving read qualities, as well as removing most adapter sequences. The exception to the latter was terminal poly-A sequences, which remained at a substantial (though reduced) prevalence throughout the pipeline. (This doesn’t seem to have caused downstream problems that I can see.)
According to FASTQC, deduplication was very effective at reducing measured duplicate levels, which for single-room samples fell from an average of 40% to 7.8% for RNA reads and from 5.5% to 0.3% for DNA reads.
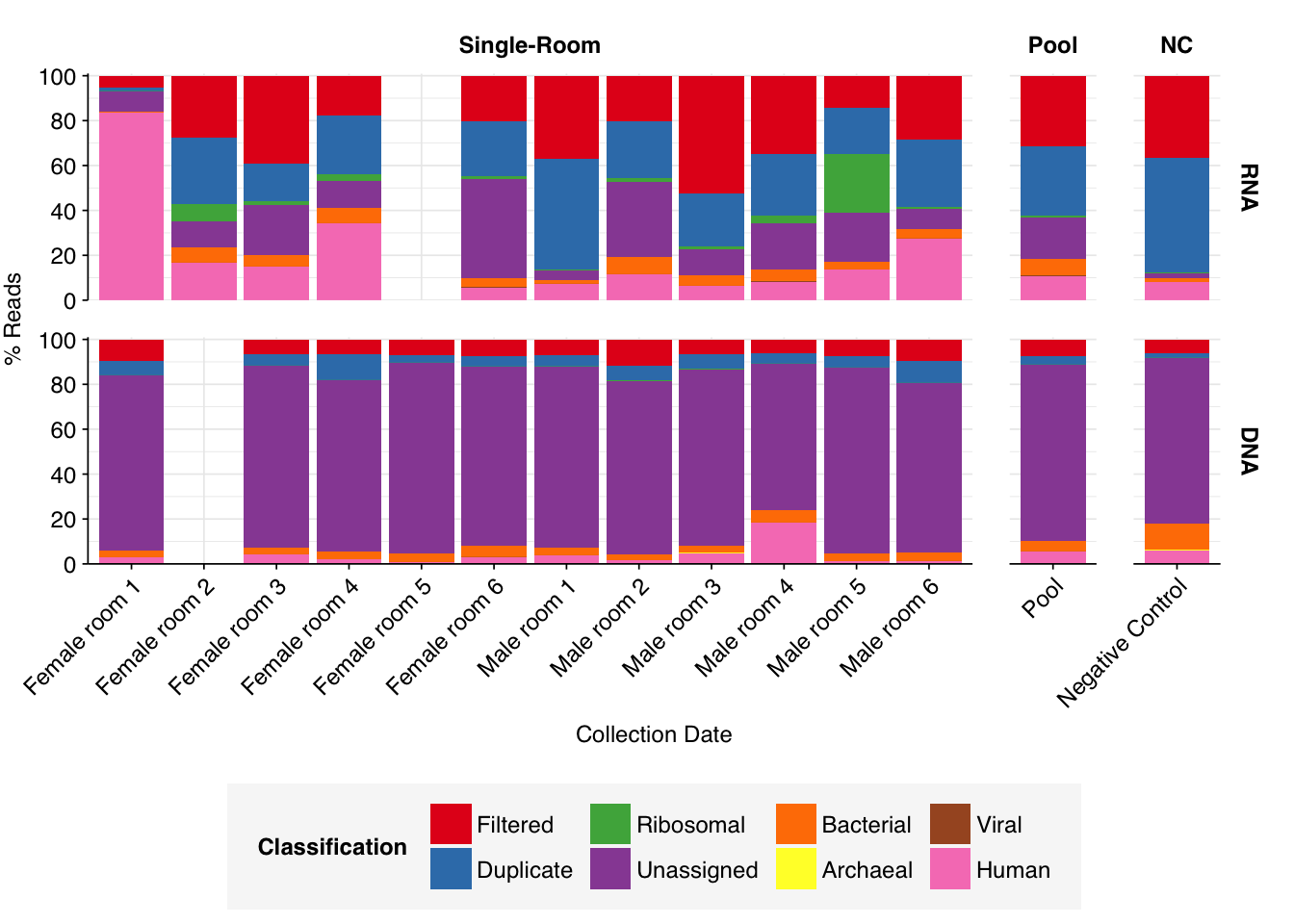
As before, to assess the high-level composition of the reads, I ran the ribodepleted files through Kraken (using the Standard 16 database) and summarized the results with Bracken. Combining these results with the read counts above gives us a breakdown of the inferred composition of the samples:
# Prepare plotting templatesg_comp_base<-ggplot(mapping=aes(x=room, y=p_reads, fill=classification))+scale_x_discrete(name="Collection Date")+facet_grid(na_type~sample_type, scales ="free", space ="free_x")+theme_rotatescale_y_pc_reads<-purrr::partial(scale_y_continuous, name ="% Reads", expand =c(0,0), labels =function(y)y*100)# Plot overall compositiong_comp<-g_comp_base+geom_col(data =read_comp_long, position ="stack")+scale_y_pc_reads(limits =c(0,1.01), breaks =seq(0,1,0.2))+scale_fill_brewer(palette ="Set1", name ="Classification")g_comp
Code
# Plot composition of minor componentsread_comp_minor<-read_comp_long%>%filter(classification%in%c("Archaeal", "Viral", "Other"))palette_minor<-brewer.pal(9, "Set1")[c(6,7,9)]g_comp_minor<-g_comp_base+geom_col(data=read_comp_minor, position ="stack")+scale_y_pc_reads()+scale_fill_manual(values=palette_minor, name ="Classification")g_comp_minor
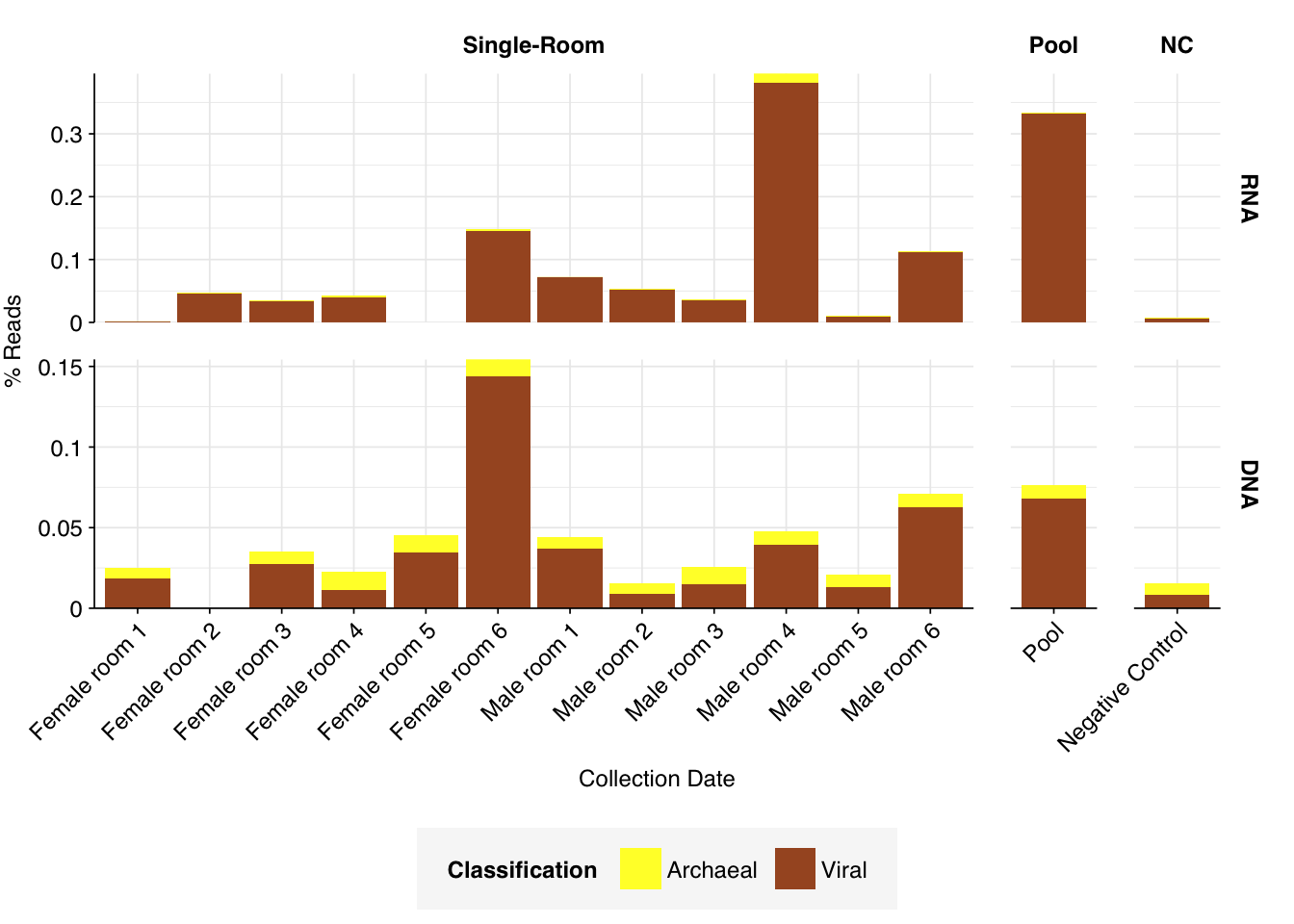
As with the Prussin data, human reads are very elevated compared to wastewater, making up an average of 21% of read pairs in RNA libraries and 4% for DNA libraries for single-room samples (in both cases, the maximum abundance of human reads across samples is much higher). Viral read abundances are higher than Prussin, averaging 0.08% for single-room RNA libraries and 0.04% for DNA libraries; for some reason, the pooled RNA library shows much higher viral prevalence, at 0.33%. The most obvious difference from the Prussin data is that DNA libraries in Rosario show a much higher fraction of non-ribosomal unassigned reads, with an average of 78% of all reads for single-room libraries.
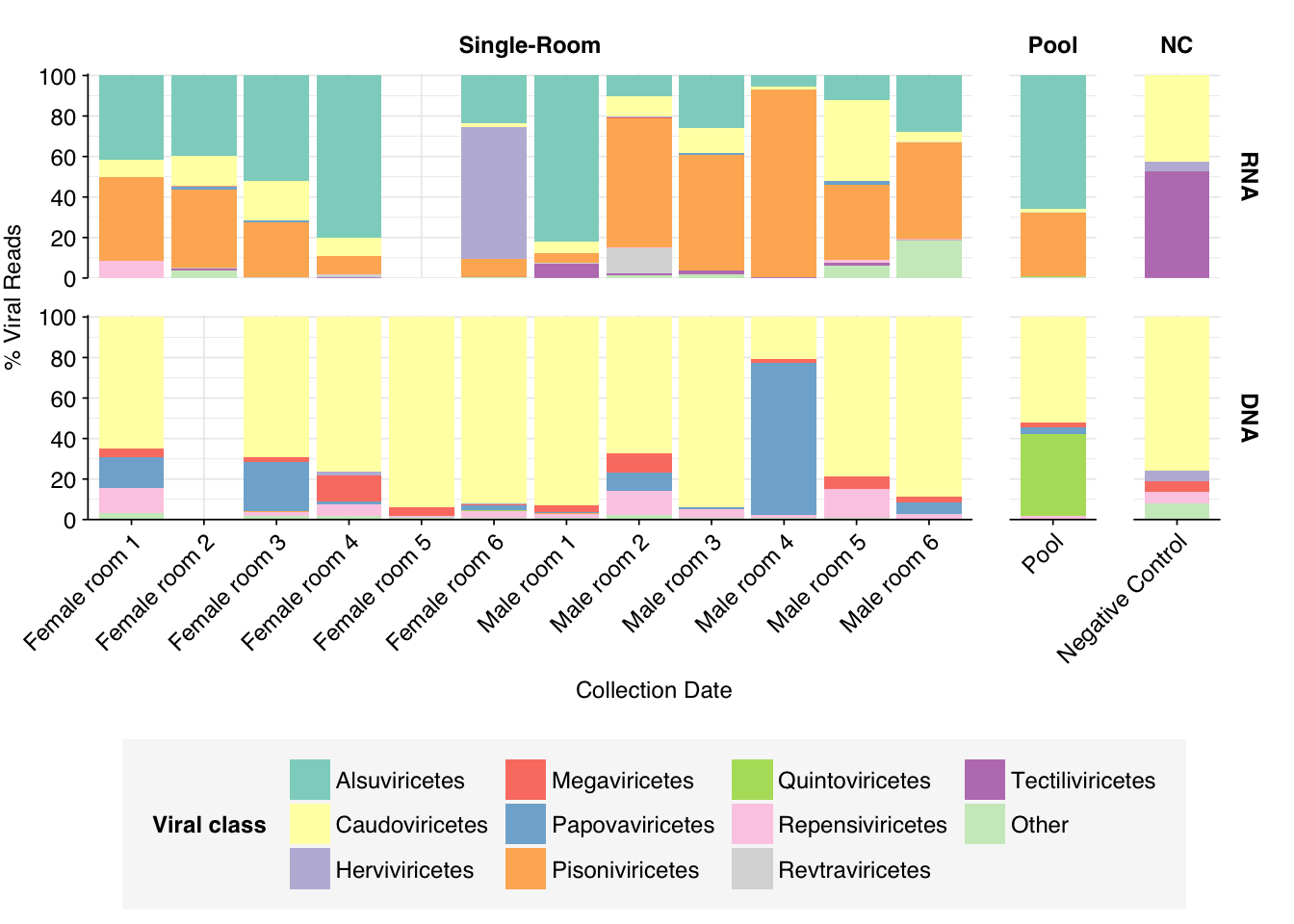
As in Prussin, viral DNA reads were dominated by Caudoviricetes phages, with many unclassifiable below the class level. Other prominent viral classes in DNA reads included Papovaviricetes (which includes Polyomaviridae and Papillomaviridae), Megaviridae (giant viruses) and Repensiviricetes (plant and fungal viruses). Pooled samples also showed a significant level of Quintoviricetes (parvoviruses).
RNA libraries, meanwhile, were dominated by Alsuviricetes (mainly plant viruses) and, notably, Pisoniviricetes (which includes coronaviruses, picornaviruses including Enterovirus, and caliciviruses including Norovirus). Caudoviricetes, Herviviricetes (herpesviruses) and Revtraviricetes (retroviruses + Hep B) were also prominent in some samples.
Of these, the strong presence of Papovaviricetes and Pisoniviricetes across multiple samples is the most interesting, as both contain important human pathogens. The presence of Quintoviricetes, Herviviricetes and Revtraviricetes in a smaller number of samples is also notable.
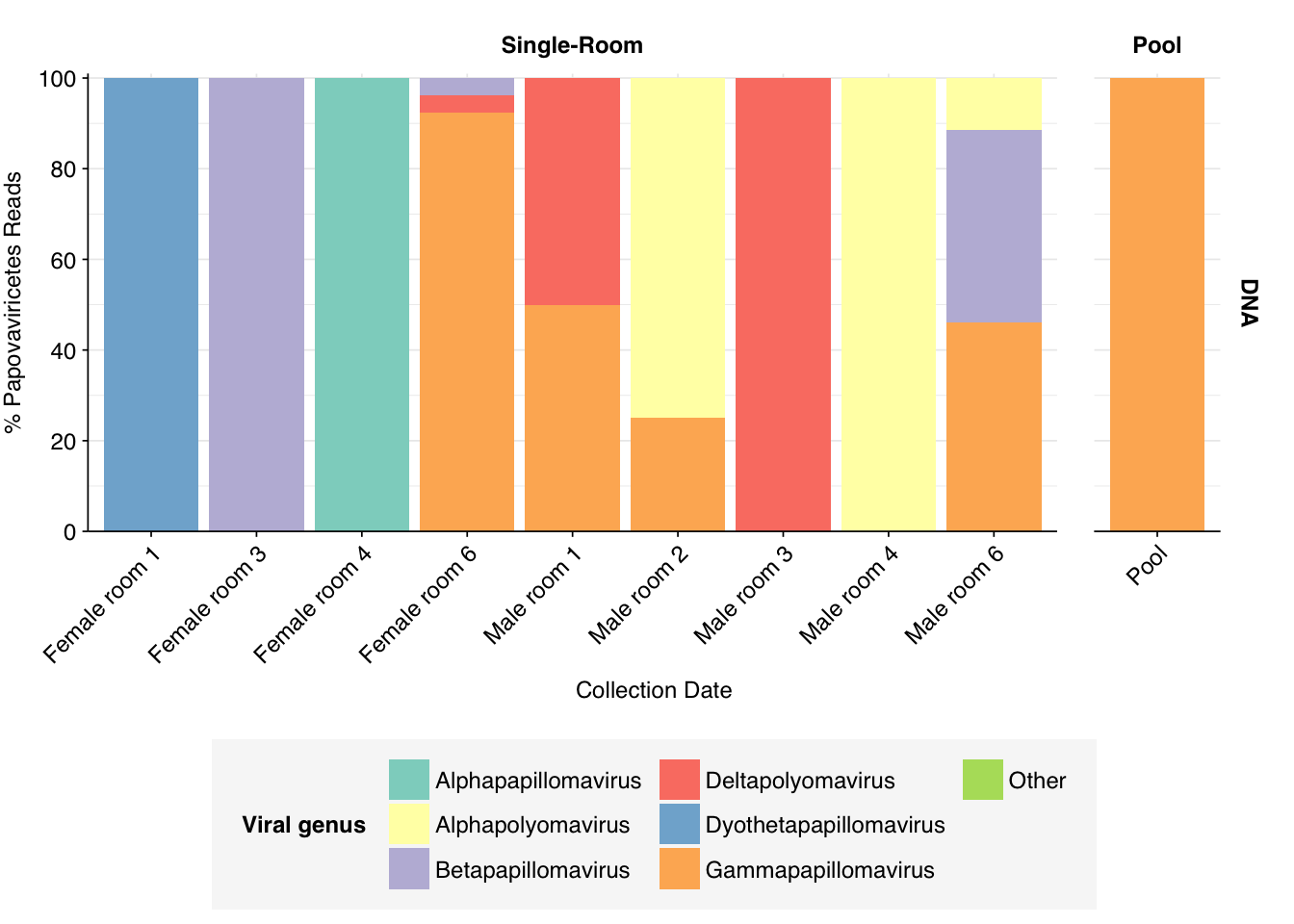
In DNA reads, Papovaviricetes are quite heterogeneous across samples, while mostly being dominated by one or two genera within samples. The three rooms with the largest Papovaviricetes fraction (Female room 1 & 3 and Male room 4) are each dominated by a single genus, specifically Dyothetapapillomavirus (of which the sole member is Dyothetapapillomavirus 1, a cat pathogen), Betapapillomavirus (a genus of human wart-causing viruses), and Alphapolyomavirus (specifically Merkel cell polyomavirus).
Code
# Get all read counts in classpapova_taxid<-2732421papova_desc_taxids_old<-papova_taxidpapova_desc_taxids_new<-unique(c(papova_desc_taxids_old, viral_taxa%>%filter(parent_taxid%in%papova_desc_taxids_old)%>%pull(taxid)))while(length(papova_desc_taxids_new)>length(papova_desc_taxids_old)){papova_desc_taxids_old<-papova_desc_taxids_newpapova_desc_taxids_new<-unique(c(papova_desc_taxids_old, viral_taxa%>%filter(parent_taxid%in%papova_desc_taxids_old)%>%pull(taxid)))}papova_counts<-kraken_reports_viral_cleaned%>%filter(taxid%in%papova_desc_taxids_new)%>%mutate(p_reads_papova =n_reads_clade/n_reads_clade[1])# Get genus compositionpapova_genera<-papova_counts%>%filter(rank=="G", na_type=="DNA")papova_genera_major_tab<-papova_genera%>%group_by(name, taxid)%>%summarize(p_reads_papova_max =max(p_reads_papova), .groups="drop")%>%filter(p_reads_papova_max>=0.04)papova_genera_major_list<-papova_genera_major_tab%>%pull(name)papova_genera_major<-papova_genera%>%filter(name%in%papova_genera_major_list)%>%select(name, taxid, sample, room, na_type, sample_type, p_reads_papova)papova_genera_minor<-papova_genera_major%>%group_by(sample, room, na_type, sample_type)%>%summarize(p_reads_papova_major =sum(p_reads_papova), .groups ="drop")%>%mutate(name ="Other", taxid=NA, p_reads_papova =1-p_reads_papova_major)%>%select(name, taxid, sample, room, na_type, sample_type, p_reads_papova)papova_genera_display<-bind_rows(papova_genera_major, papova_genera_minor)%>%arrange(desc(p_reads_papova))%>%mutate(name =factor(name, levels=c(papova_genera_major_list, "Other")))%>%rename(p_reads =p_reads_papova, classification=name)# Plotg_papova_genera<-g_comp_base+geom_col(data=papova_genera_display, position ="stack")+scale_y_continuous(name="% Papovaviricetes Reads", limits=c(0,1.01), breaks =seq(0,1,0.2), expand=c(0,0), labels =function(y)y*100)+scale_fill_manual(values=palette_viral, name ="Viral genus")+guides(fill=guide_legend(ncol=3))g_papova_genera
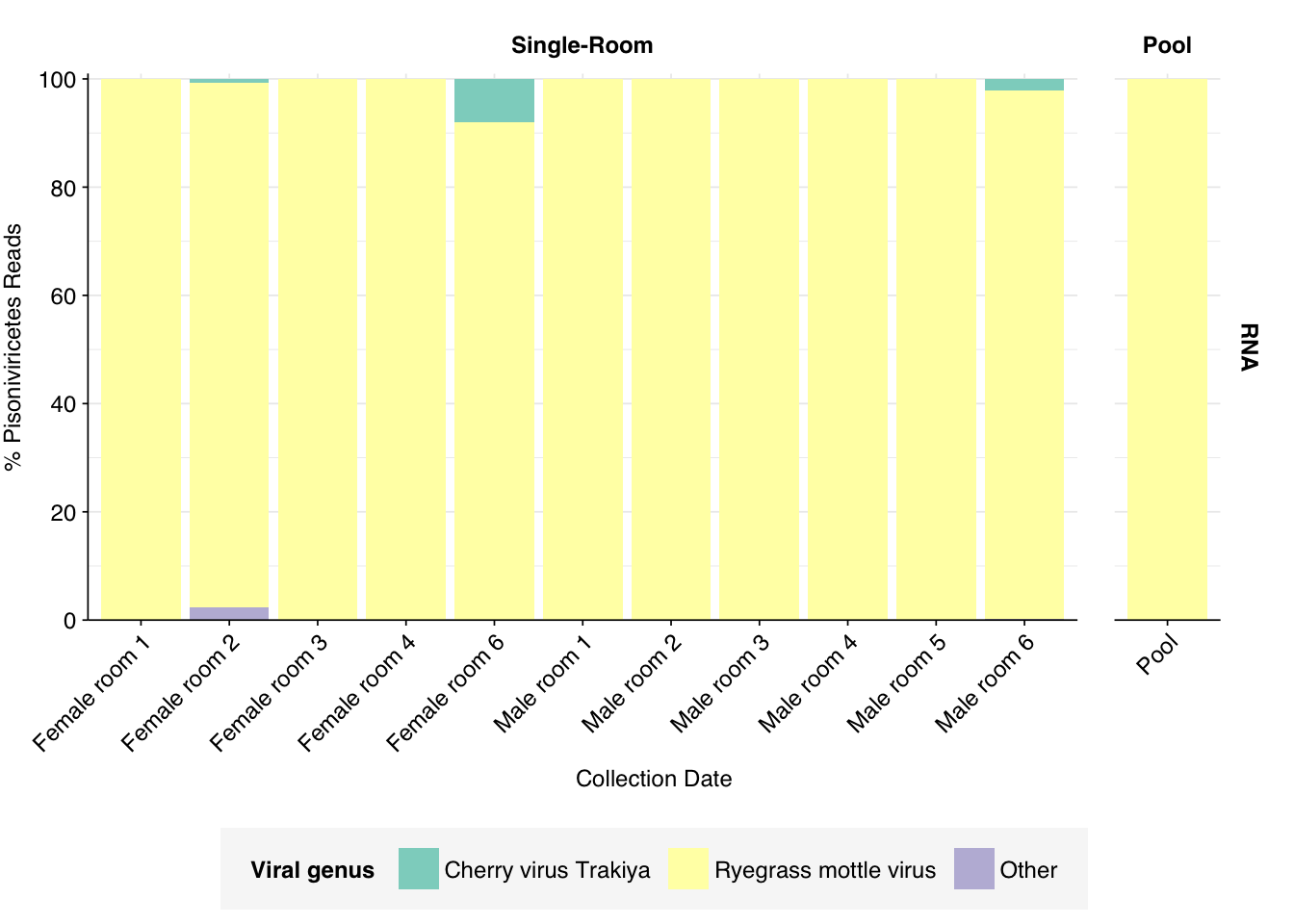
In RNA reads, Pisoniviricetes are conversely dominated by a single genus, Sobemovirus, across all samples, and specifically a single species. Disappointingly, this is Ryegrass mottle virus, a plant pathogen.
Code
# Get all read counts in classpisoni_taxid<-2732506pisoni_desc_taxids_old<-pisoni_taxidpisoni_desc_taxids_new<-unique(c(pisoni_desc_taxids_old, viral_taxa%>%filter(parent_taxid%in%pisoni_desc_taxids_old)%>%pull(taxid)))while(length(pisoni_desc_taxids_new)>length(pisoni_desc_taxids_old)){pisoni_desc_taxids_old<-pisoni_desc_taxids_newpisoni_desc_taxids_new<-unique(c(pisoni_desc_taxids_old, viral_taxa%>%filter(parent_taxid%in%pisoni_desc_taxids_old)%>%pull(taxid)))}pisoni_counts<-kraken_reports_viral_cleaned%>%filter(taxid%in%pisoni_desc_taxids_new)%>%mutate(p_reads_pisoni =n_reads_clade/n_reads_clade[1])# Get genus compositionpisoni_genera<-pisoni_counts%>%filter(rank=="S", na_type=="RNA")pisoni_genera_major_tab<-pisoni_genera%>%group_by(name, taxid)%>%summarize(p_reads_pisoni_max =max(p_reads_pisoni), .groups="drop")%>%filter(p_reads_pisoni_max>=0.04)pisoni_genera_major_list<-pisoni_genera_major_tab%>%pull(name)pisoni_genera_major<-pisoni_genera%>%filter(name%in%pisoni_genera_major_list)%>%select(name, taxid, sample, room, na_type, sample_type, p_reads_pisoni)pisoni_genera_minor<-pisoni_genera_major%>%group_by(sample, room, na_type, sample_type)%>%summarize(p_reads_pisoni_major =sum(p_reads_pisoni), .groups ="drop")%>%mutate(name ="Other", taxid=NA, p_reads_pisoni =1-p_reads_pisoni_major)%>%select(name, taxid, sample, room, na_type, sample_type, p_reads_pisoni)pisoni_genera_display<-bind_rows(pisoni_genera_major, pisoni_genera_minor)%>%arrange(desc(p_reads_pisoni))%>%mutate(name =factor(name, levels=c(pisoni_genera_major_list, "Other")))%>%rename(p_reads =p_reads_pisoni, classification=name)# Plotg_pisoni_genera<-g_comp_base+geom_col(data=pisoni_genera_display, position ="stack")+scale_y_continuous(name="% Pisoniviricetes Reads", limits=c(0,1.01), breaks =seq(0,1,0.2), expand=c(0,0), labels =function(y)y*100)+scale_fill_manual(values=palette_viral, name ="Viral genus")+guides(fill=guide_legend(ncol=3))g_pisoni_genera
Human-infecting virus reads: validation
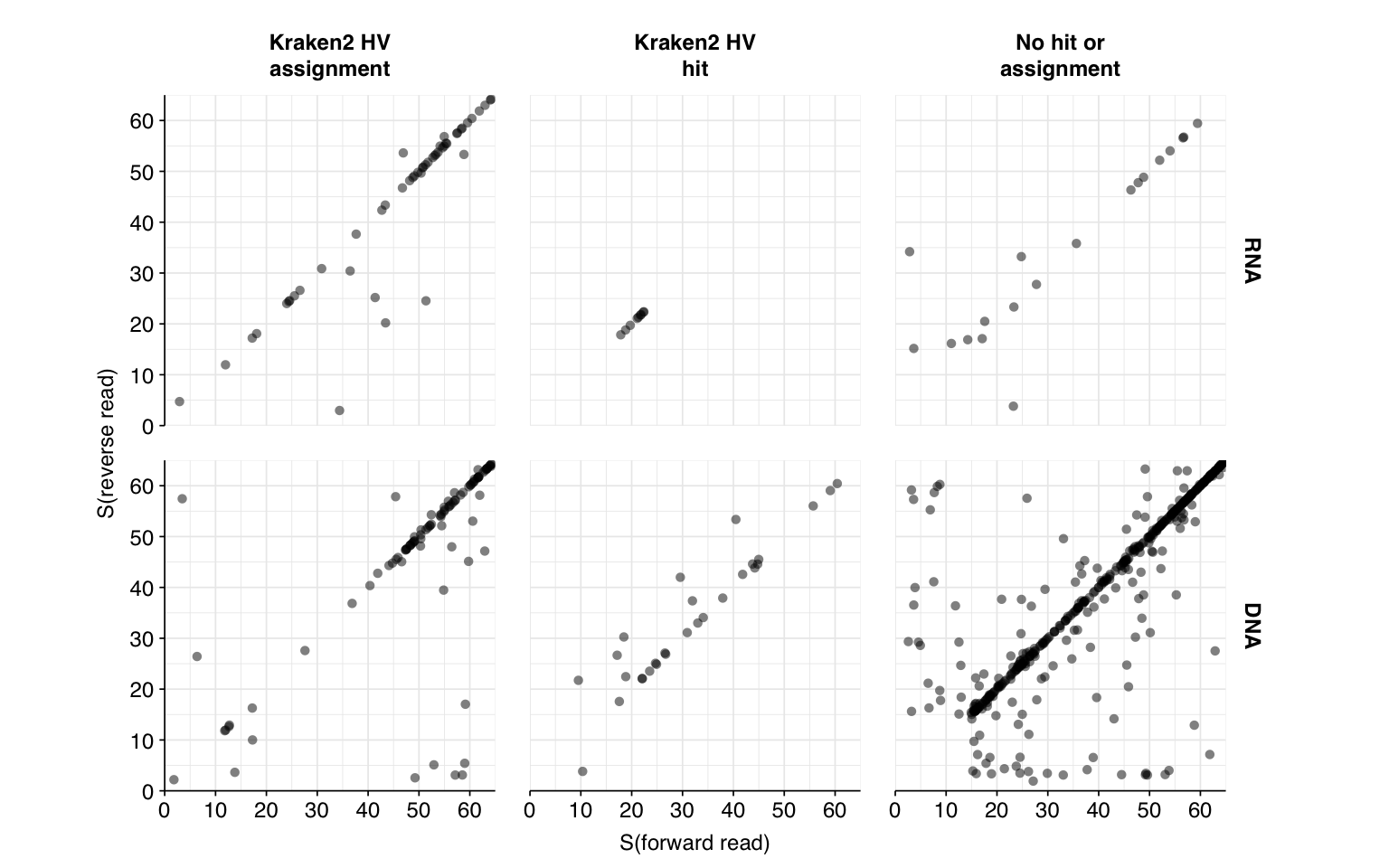
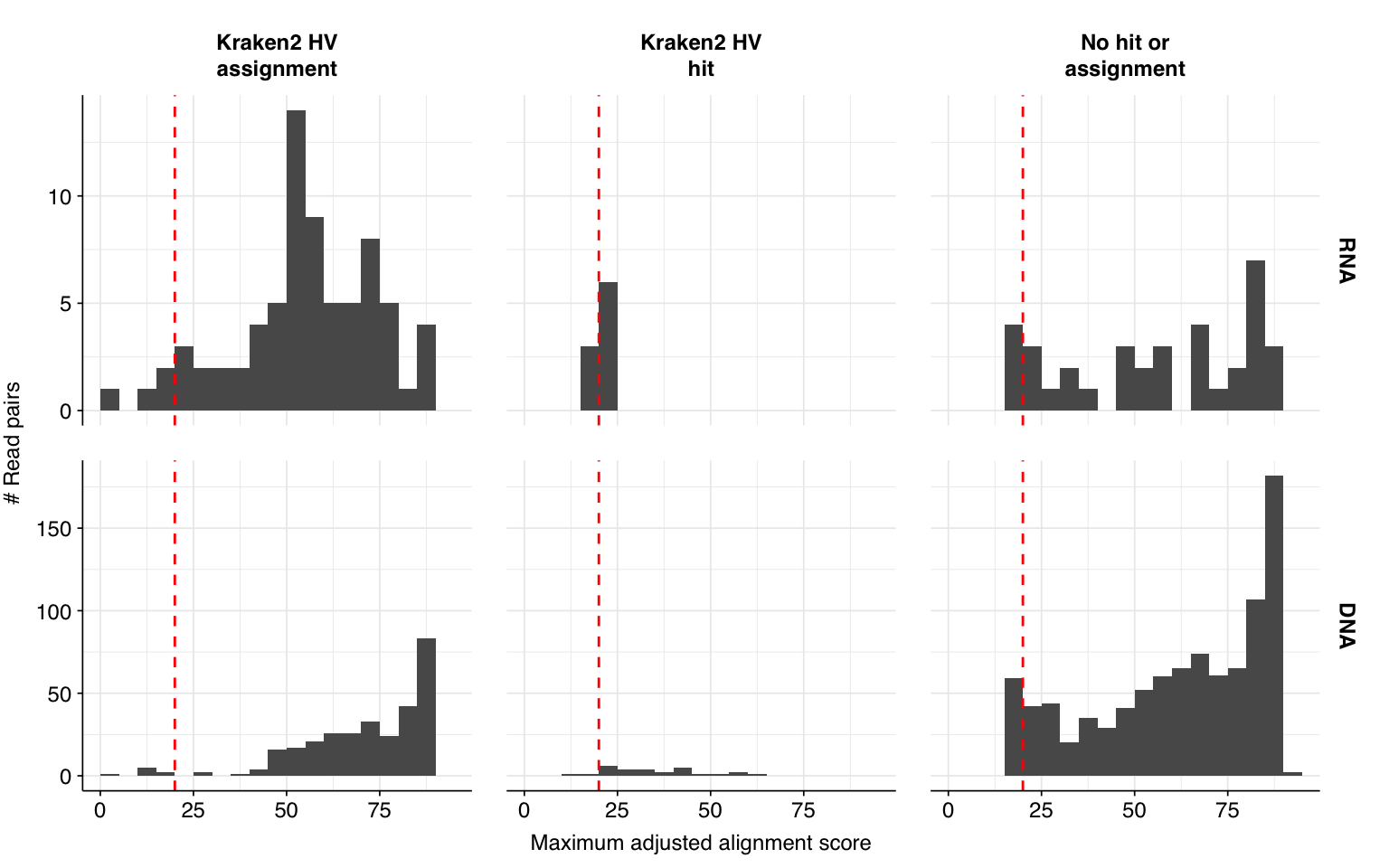
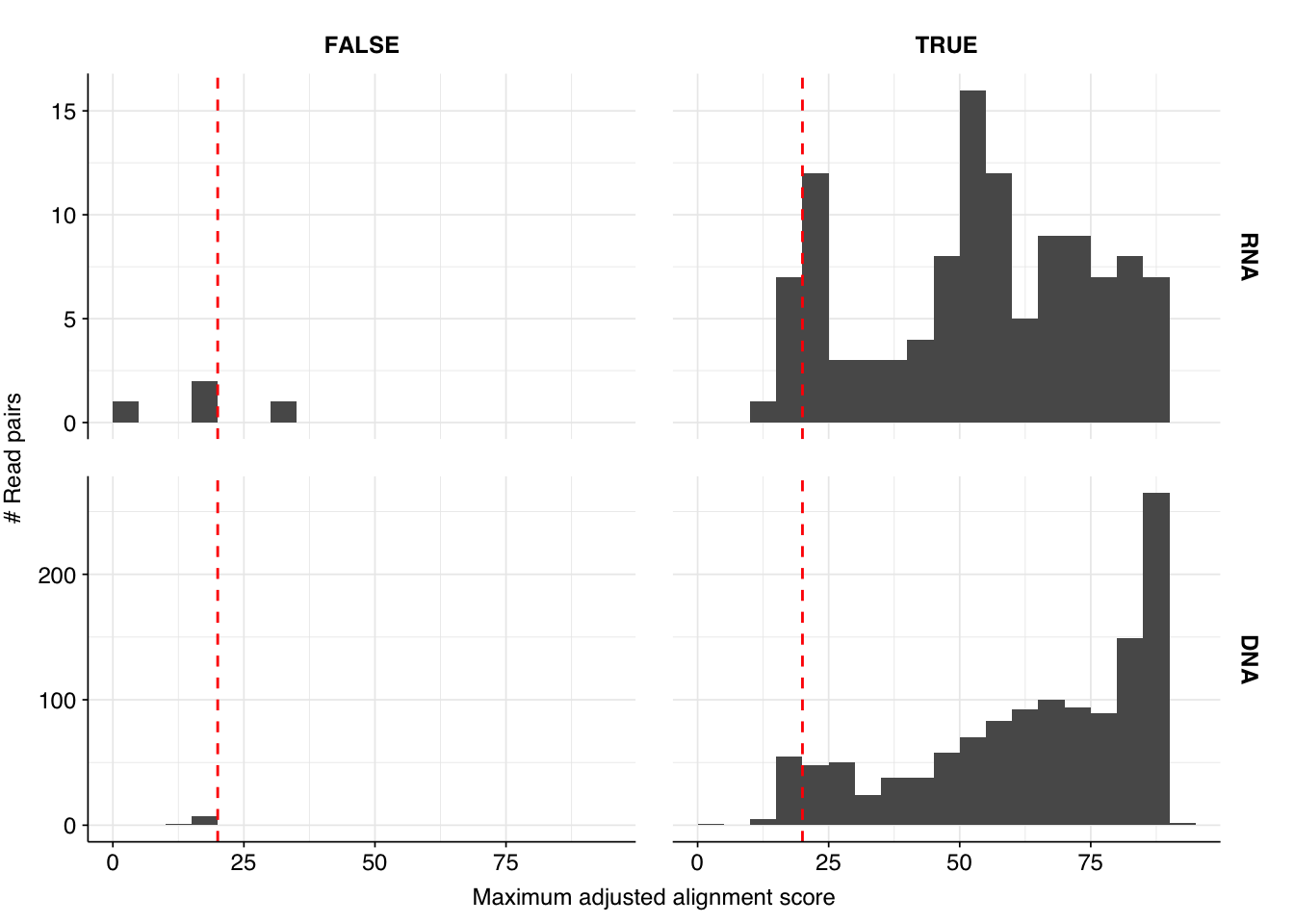
Next, I investigated the human-infecting virus read content of these unenriched samples. Using the same workflow I used for Prussin et al, I identified 118 RNA read pairs and 1269 DNA read pairs as putatively human viral: 0.004% and 0.017% of surviving reads, respectively.
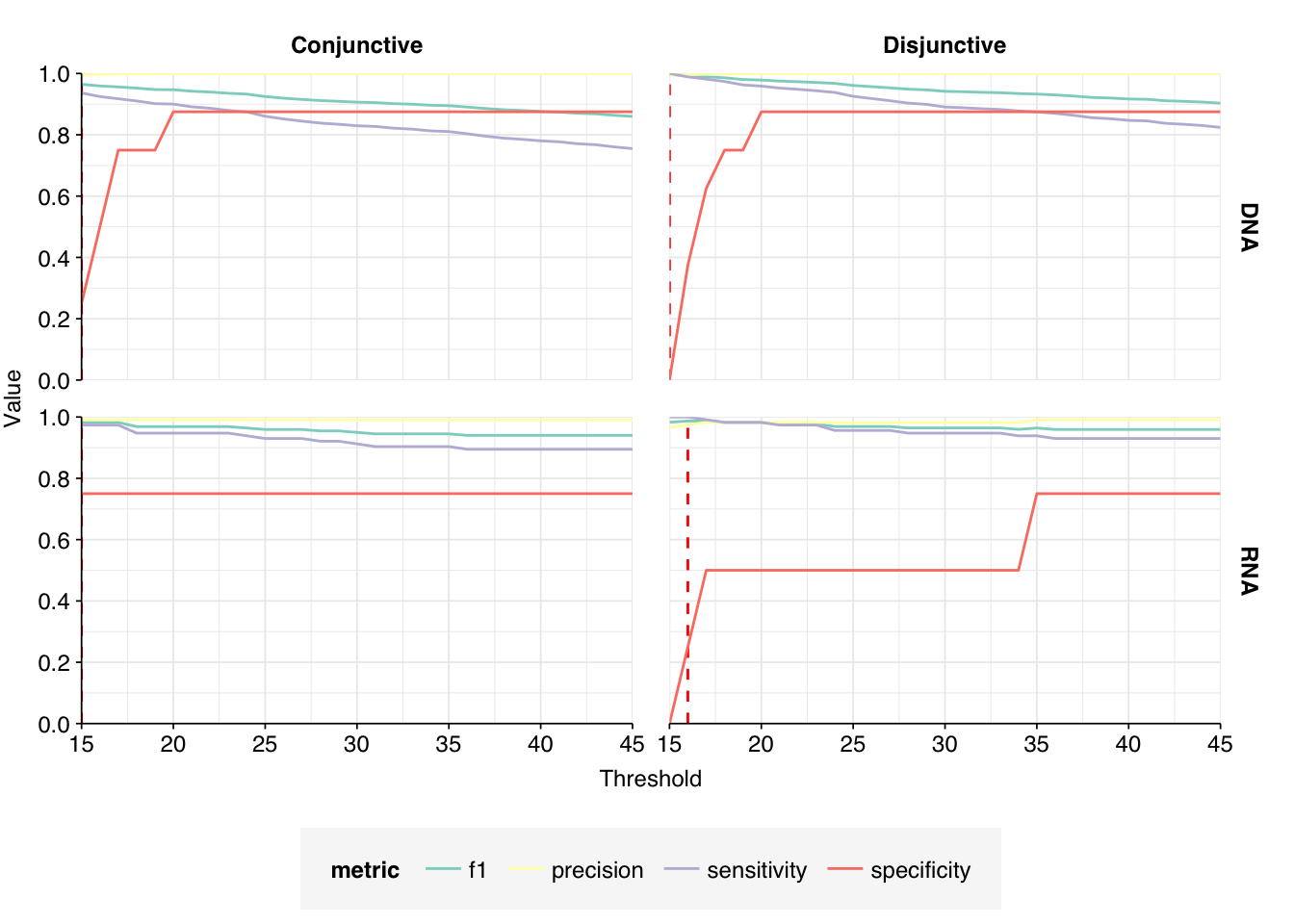
These results look good on visual inspection, and indeed precision and sensitivity are both very high. For a disjunctive score threshold of 20, my updated workflow achieves an F1 score of 98.2% for RNA sequences and 97.9% for DNA sequences.
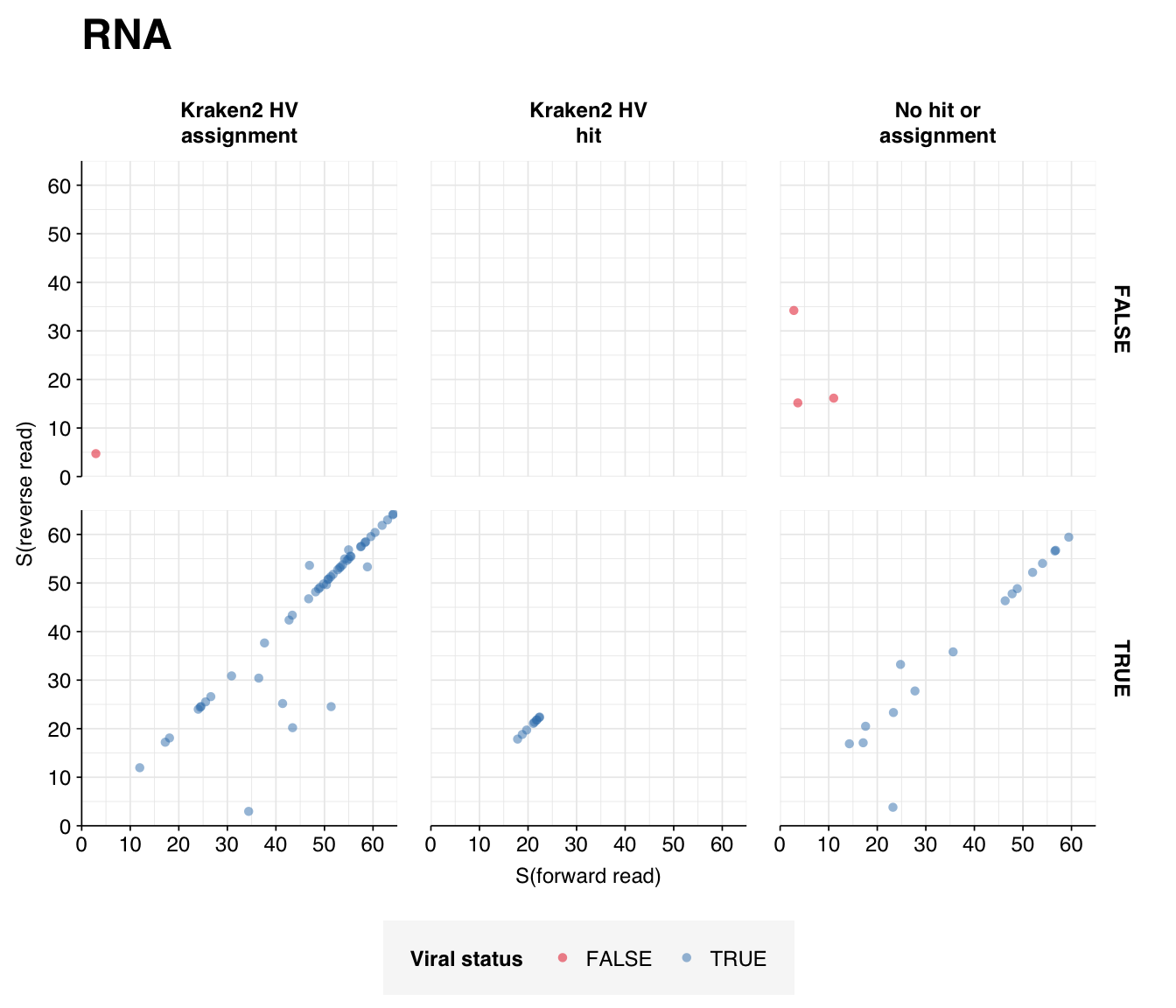
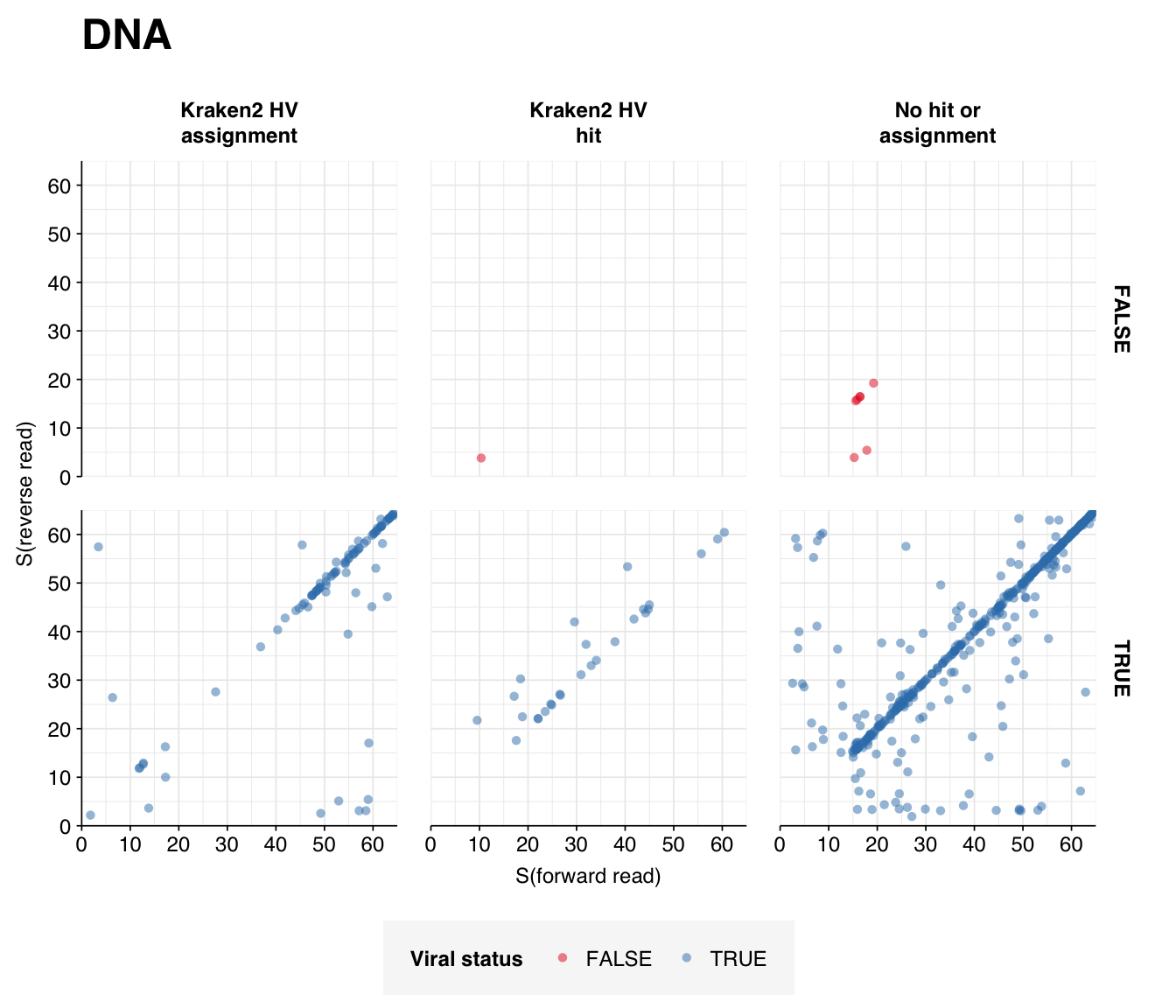
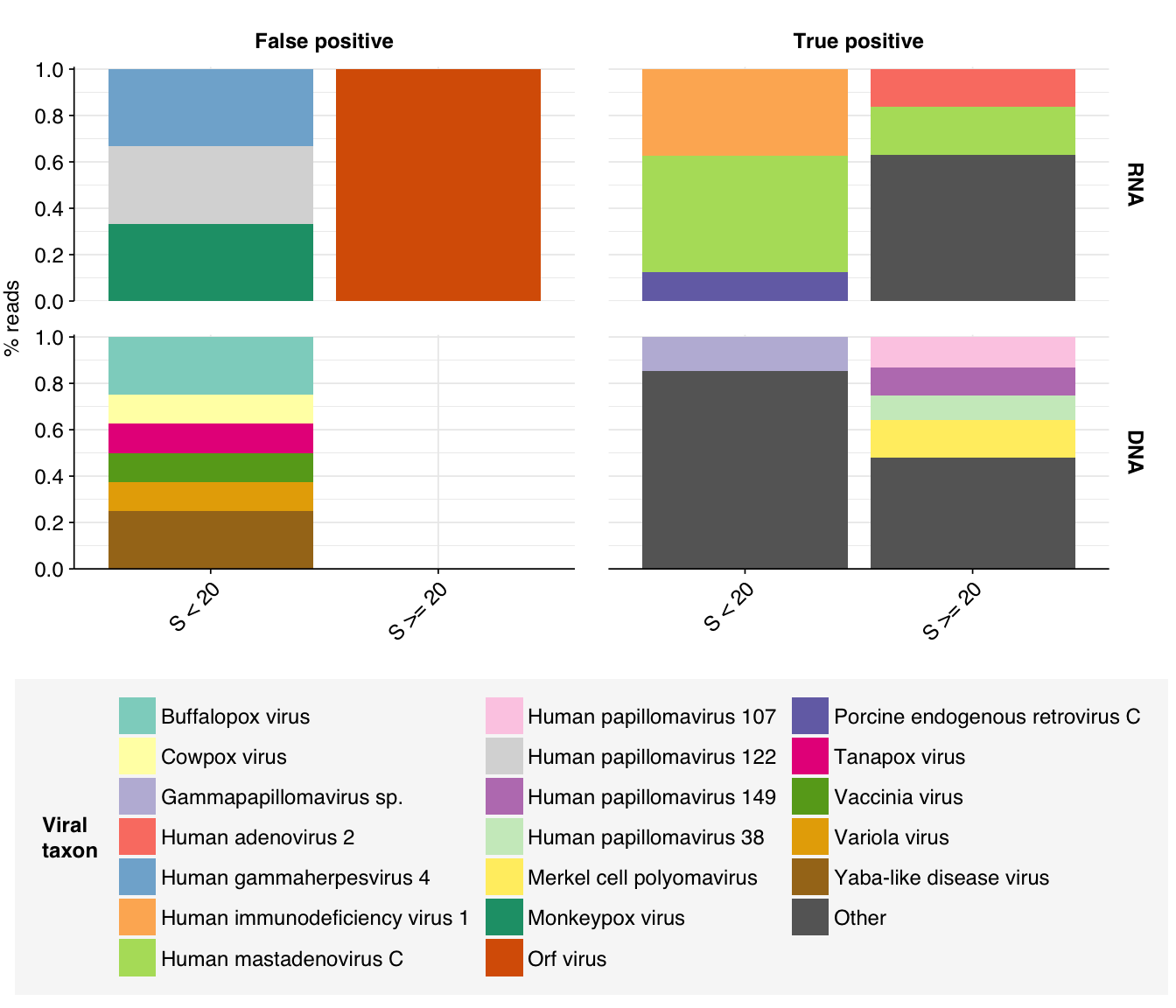
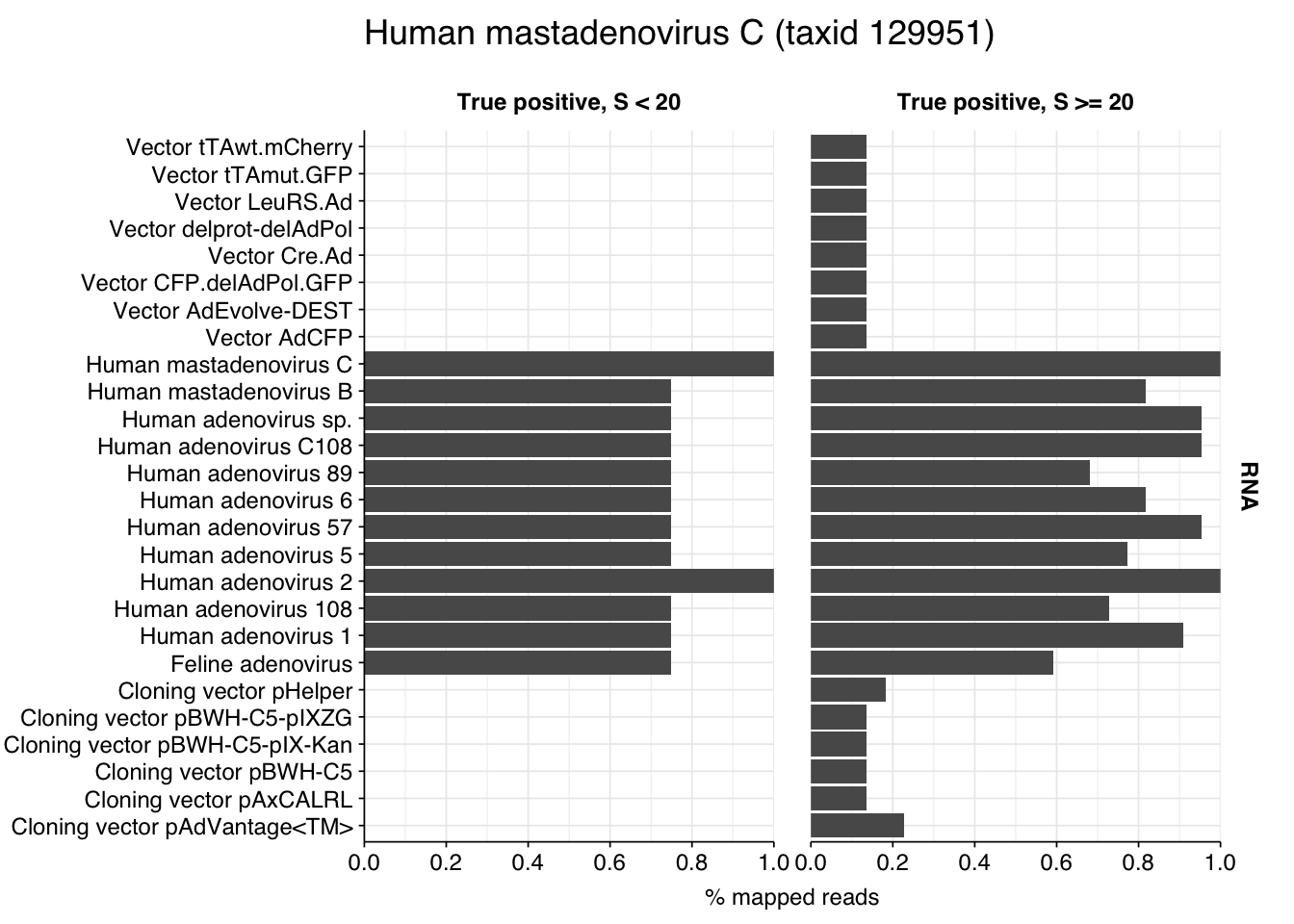
Looking into the composition of different read groups, the most notable observations for me are the predominance of (1) HIV reads among low-scoring RNA “true-positives”, and (2) human mastadenovirus C among RNA true-positives in general:
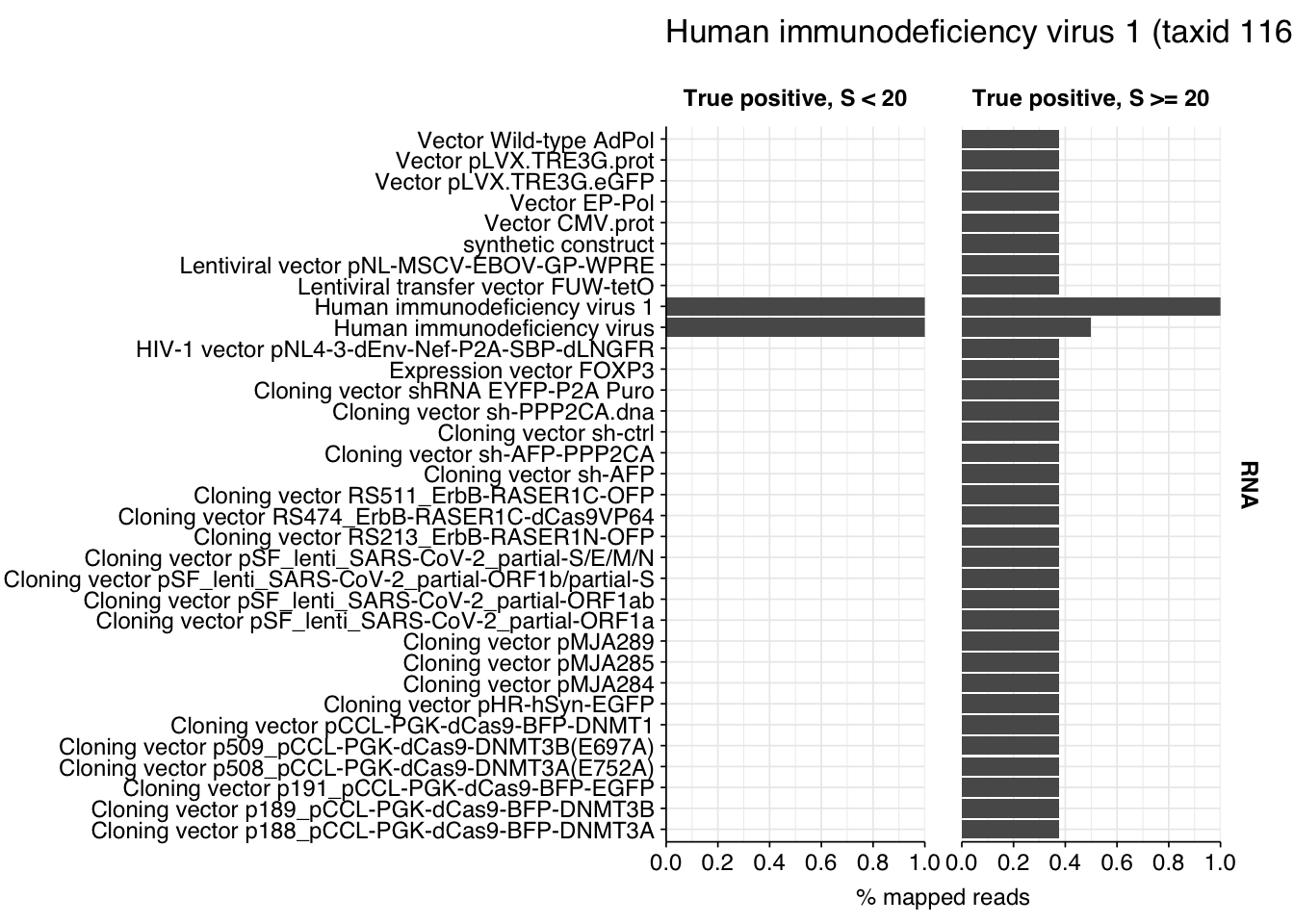
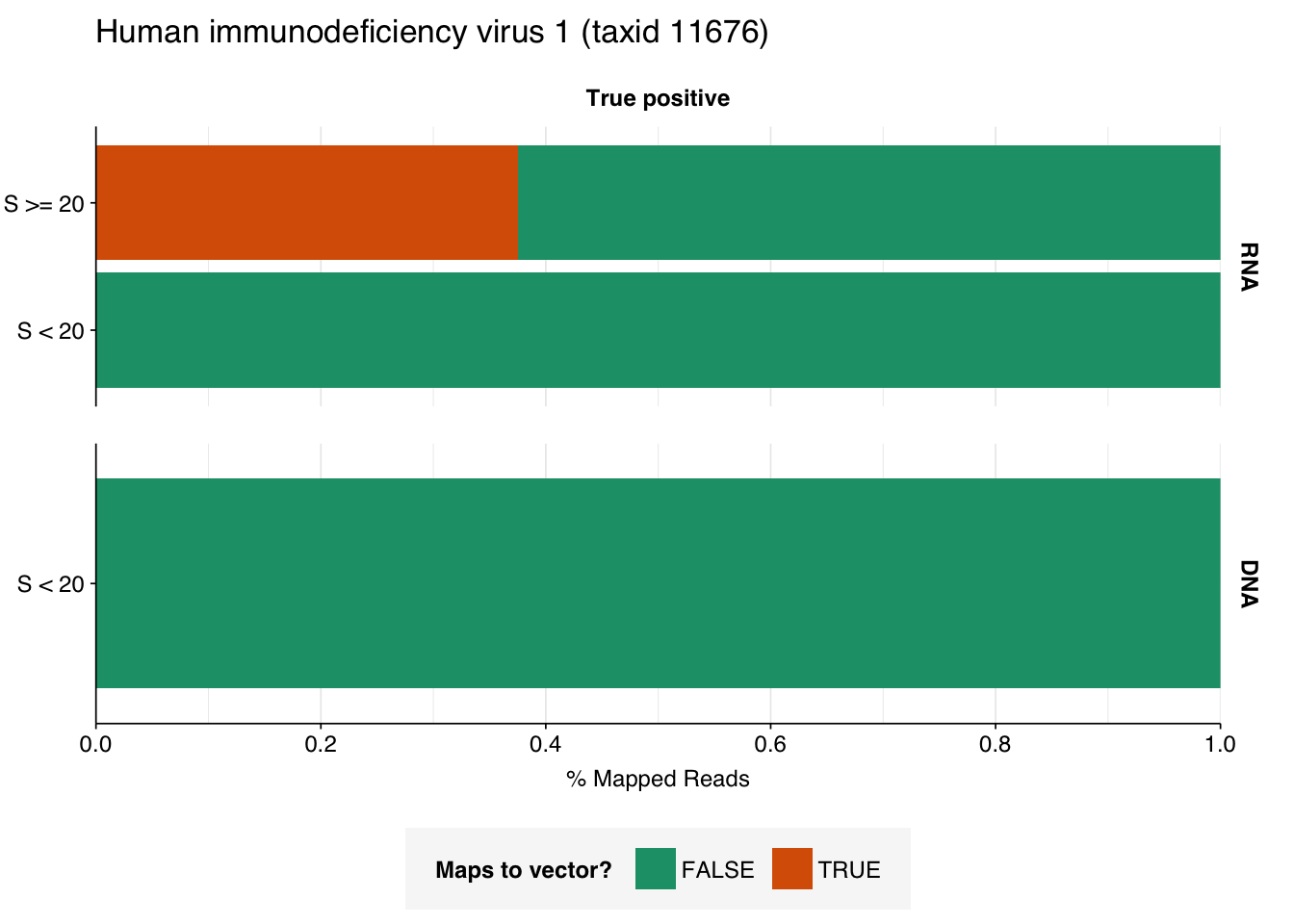
There are only eight low-scoring true-positive RNA reads, three of which are HIV. These do seem to be real; at least, they don’t BLAST to anything else. Ironically the eight high-scoring “true-positive” HIV reads seem more likely to be fake; they also map to a range of cloning vectors:
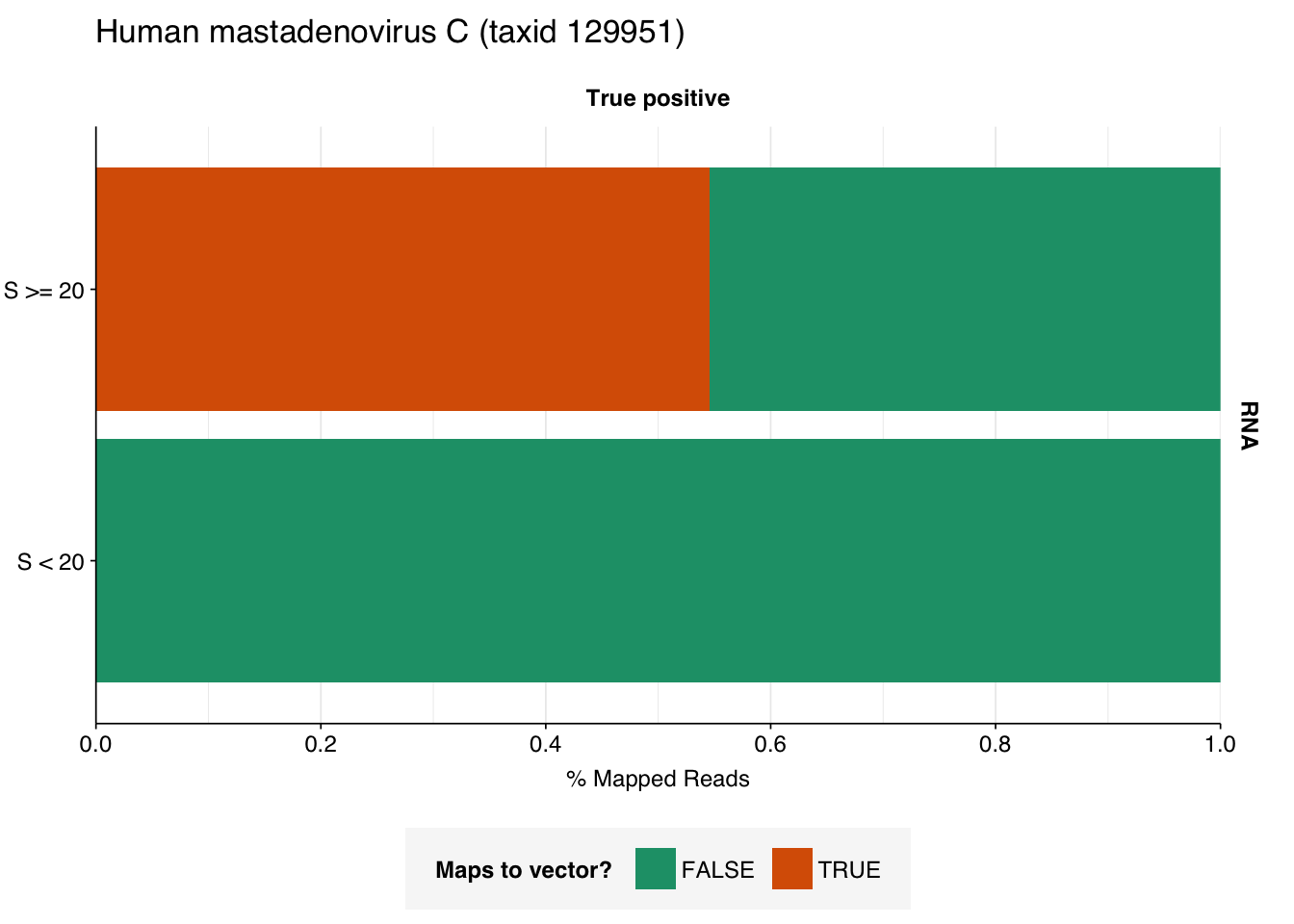
A similar pattern is seen for the Human mastadenovirus C hits: low-scoring true-positives only map to adenoviruses, but high-scoring ones also map to a range of cloning vectors.
The plots above only tell us the total number of reads that BLAST to each cloning vector; they don’t tell us how many reads map to any vector. In particular, they can’t distinguish between a situation where many reads map to a few cloning vectors, and one where a few reads map to many. This turns out to be about 35% of high-scoring HIV RNA reads and 55% of high-scoring mastadenovirus RNA reads:
This is more concerning for the mastadenovirus reads, which make up a nontrivial fraction of all high-scoring RNA “true-positives”. As in my last post, though, it’s hard to know what to take from this; many vectors are based on lentiviral or adenoviral backbones, and it’s not always easy to distinguish between true virus and virus-based vector. I’m going to stick with my pipeline as it is for now, but it’s worth keeping this in mind in interpreting the results.
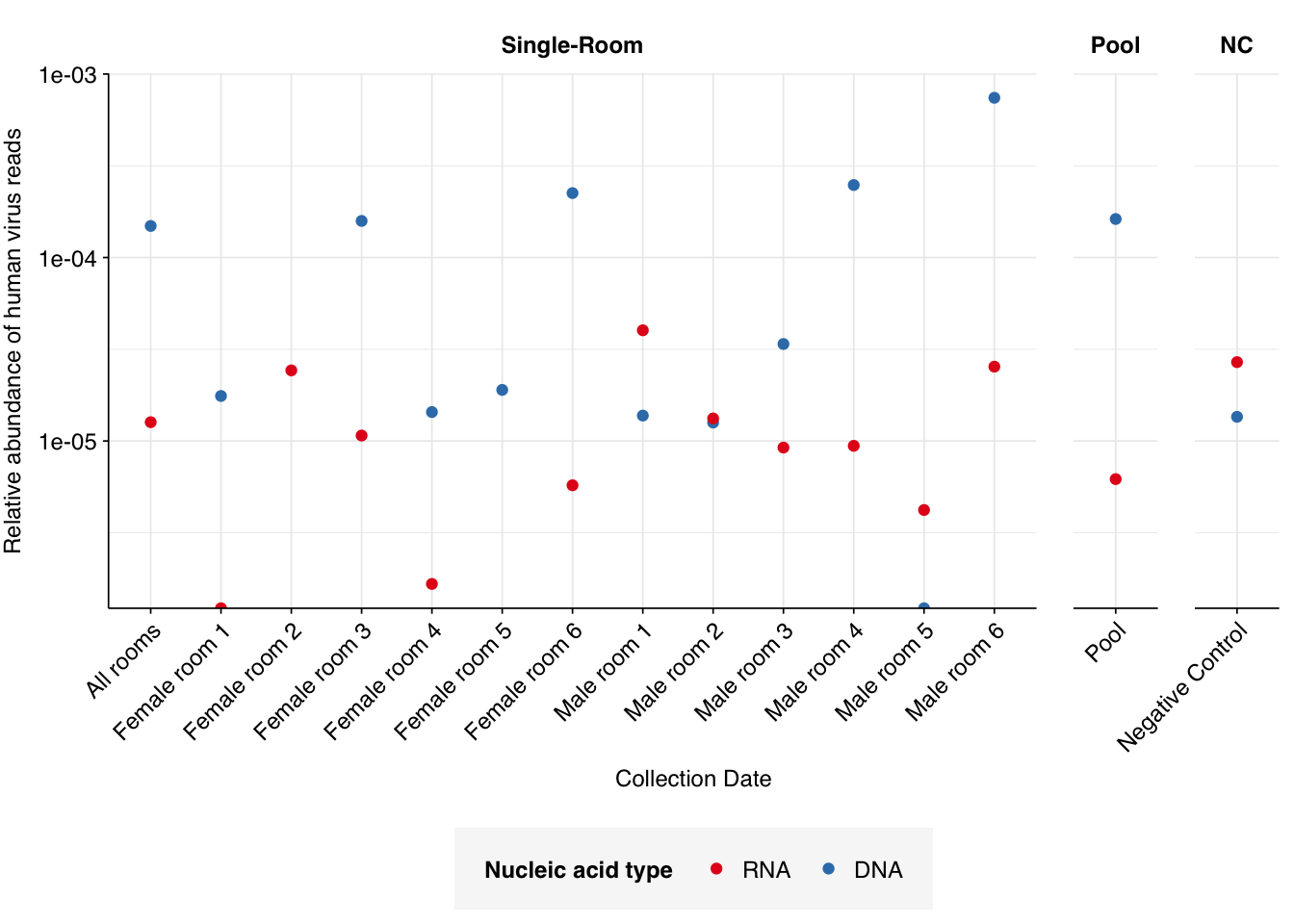
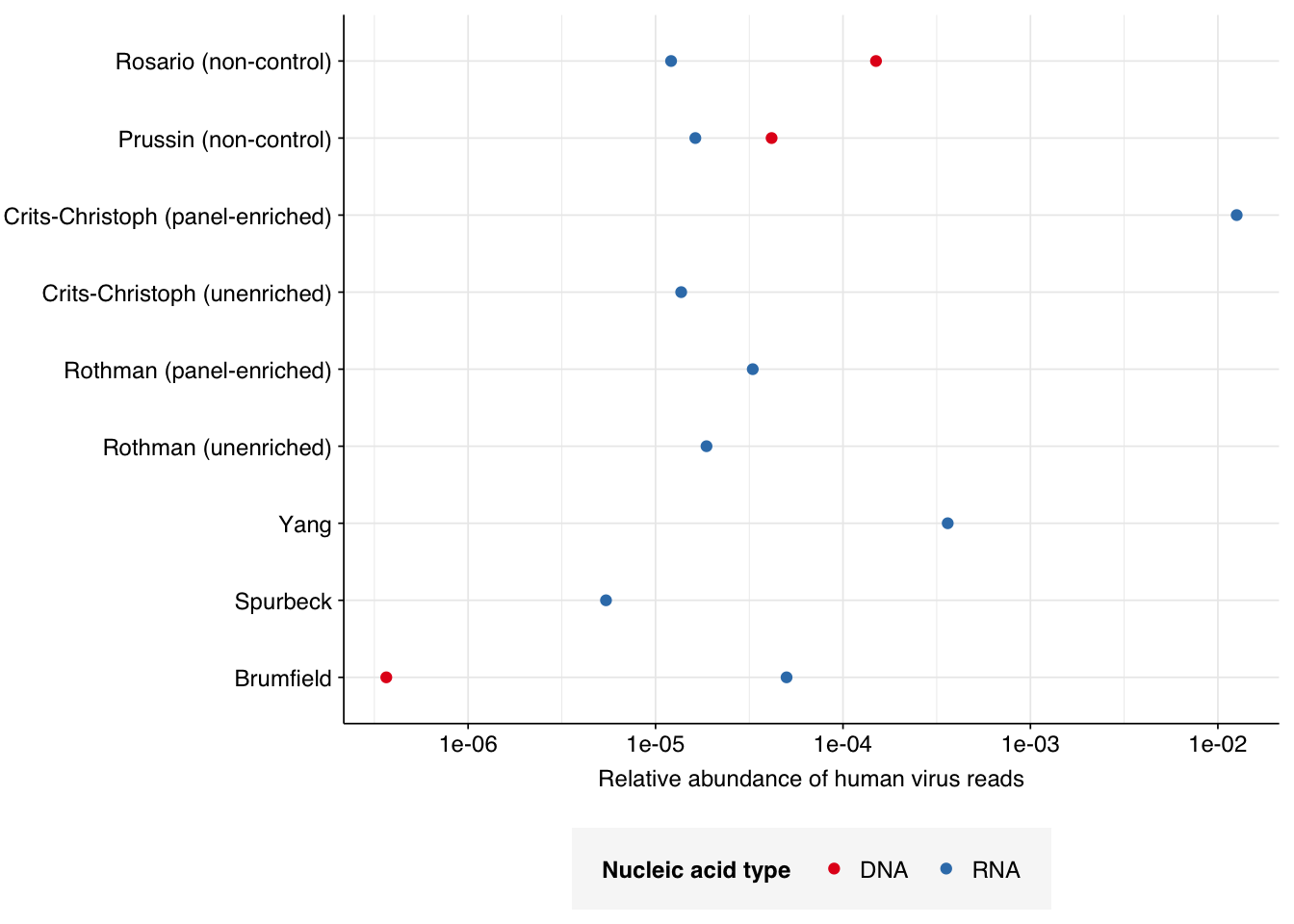
In non-control samples, applying a disjunctive cutoff at S=20 identifies 97 RNA reads and 1204 DNA reads as human-viral. This gives an overall relative HV abundance of \(1.21 \times 10^{-5}\) for RNA reads and \(1.50 \times 10^{-4}\) for DNA reads. For DNA reads, HV RA in the negative controls is about 10x lower than the non-control samples; however, for RNA reads, the HV RA in the negative controls is actually higher than in the non-controls. This, combined with the very low absolute read count, suggests that we shouldn’t take the HV results for RNA reads too seriously.
Code
# Visualizeg_phv_agg<-ggplot(read_counts_agg, aes(x=room, color=na_type))+geom_point(aes(y=p_reads_hv))+scale_y_log10("Relative abundance of human virus reads")+scale_x_discrete(name="Collection Date")+facet_grid(.~sample_type, scales ="free", space ="free_x")+scale_color_na()+theme_rotateg_phv_agg
I’m going to focus on the DNA reads here, since as discussed above I expect the RNA reads to be noisy and not very informative – which seems to in fact be the case.
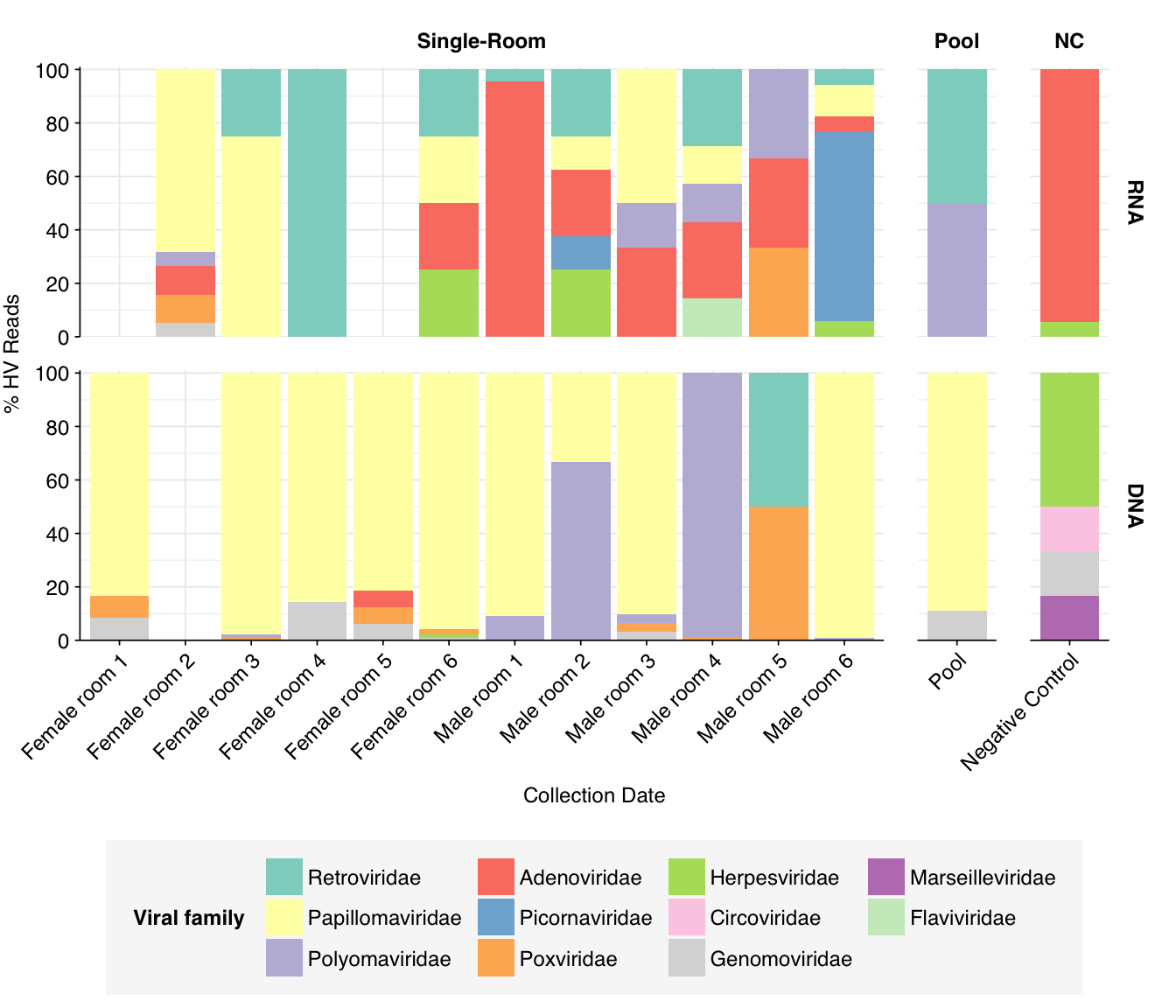
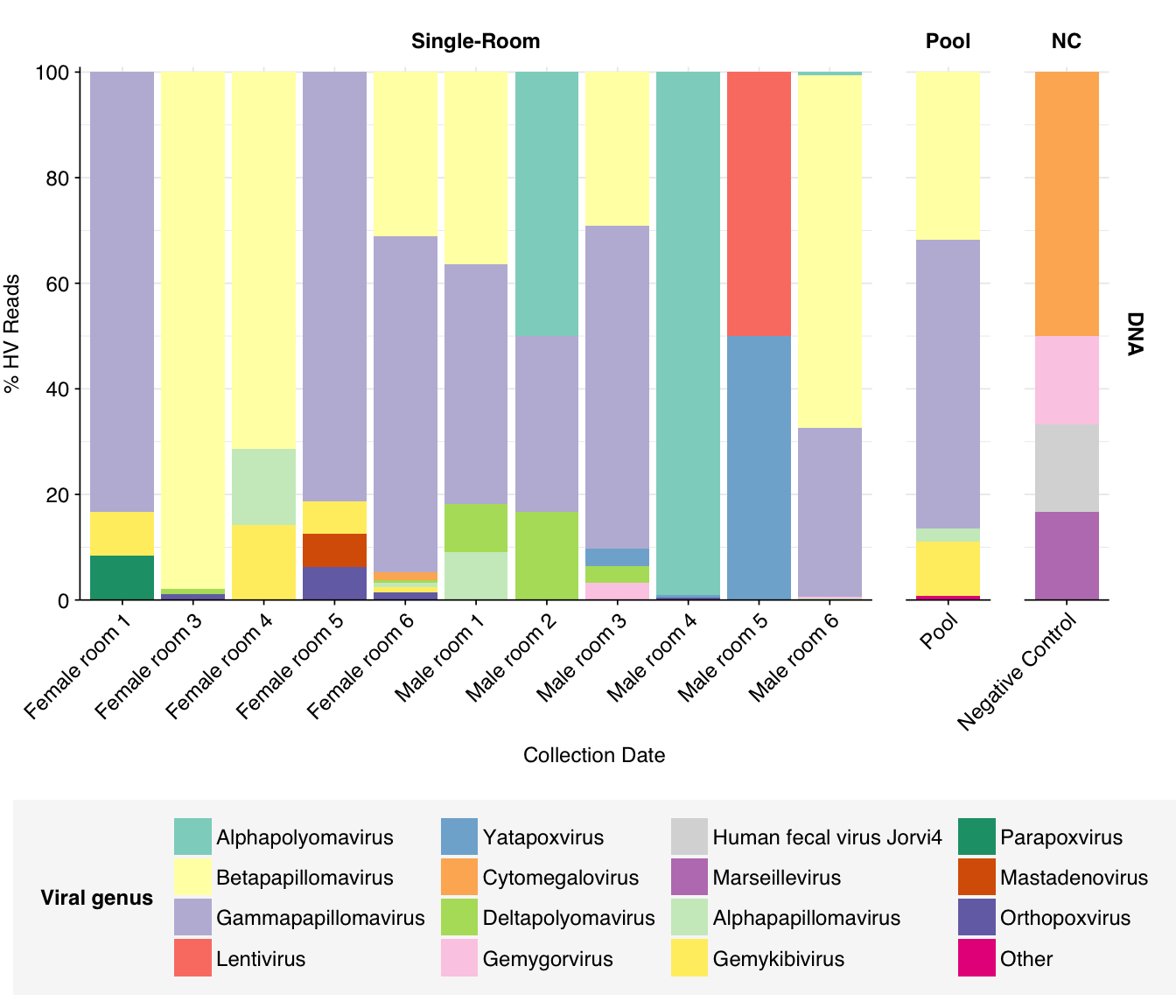
At the family level, as in Prussin, we see that Papillomaviridae, and Polyomaviridae dominate DNA reads in most samples, with Poxviridae and Genomoviridae showing significant presence in several samples. The first of these is primarily Betapapillomavirus and Gammapapillomavirus, with some Alphapapillomavirus; the second is primarily Alphapolyomavirus and Deltapolyomavirus. All of these are made up of several different viruses at the species level. Genomoviridae is represented primarily by Gemykibivirus, while Poxviridae is represented by small numbers of reads from several species, including vaccinia, cowpox and tanapox. (There’s also one variola read, which would be alarming, but BLASTN thinks it isn’t real.)
Compared to Prussin, the most striking difference is the absence of herpesvirus reads; whereas HCMV was dominant in the Prussin data, it barely shows up here except in the controls.
Code
# Get viral taxon names for putative HV readsviral_taxa$name[viral_taxa$taxid==249588]<-"Mamastrovirus"viral_taxa$name[viral_taxa$taxid==194960]<-"Kobuvirus"viral_taxa$name[viral_taxa$taxid==688449]<-"Salivirus"viral_taxa$name[viral_taxa$taxid==585893]<-"Picobirnaviridae"viral_taxa$name[viral_taxa$taxid==333922]<-"Betapapillomavirus"viral_taxa$name[viral_taxa$taxid==334207]<-"Betapapillomavirus 3"viral_taxa$name[viral_taxa$taxid==369960]<-"Porcine type-C oncovirus"viral_taxa$name[viral_taxa$taxid==333924]<-"Betapapillomavirus 2"viral_taxa$name[viral_taxa$taxid==687329]<-"Anelloviridae"viral_taxa$name[viral_taxa$taxid==325455]<-"Gammapapillomavirus"viral_taxa$name[viral_taxa$taxid==333750]<-"Alphapapillomavirus"viral_taxa$name[viral_taxa$taxid==694002]<-"Betacoronavirus"viral_taxa$name[viral_taxa$taxid==334202]<-"Mupapillomavirus"viral_taxa$name[viral_taxa$taxid==197911]<-"Alphainfluenzavirus"viral_taxa$name[viral_taxa$taxid==186938]<-"Respirovirus"viral_taxa$name[viral_taxa$taxid==333926]<-"Gammapapillomavirus 1"viral_taxa$name[viral_taxa$taxid==337051]<-"Betapapillomavirus 1"viral_taxa$name[viral_taxa$taxid==337043]<-"Alphapapillomavirus 4"viral_taxa$name[viral_taxa$taxid==694003]<-"Betacoronavirus 1"viral_taxa$name[viral_taxa$taxid==334204]<-"Mupapillomavirus 2"viral_taxa$name[viral_taxa$taxid==334208]<-"Betapapillomavirus 4"viral_taxa$name[viral_taxa$taxid==333928]<-"Gammapapillomavirus 2"viral_taxa$name[viral_taxa$taxid==337039]<-"Alphapapillomavirus 2"mrg_hv_named<-mrg_hv%>%left_join(viral_taxa, by="taxid")# Discover viral species & genera for HV readsraise_rank<-function(read_db, taxid_db, out_rank="species", verbose=FALSE){# Get higher ranks than search rankranks<-c("subspecies", "species", "subgenus", "genus", "subfamily", "family", "suborder", "order", "class", "subphylum", "phylum", "kingdom", "superkingdom")rank_match<-which.max(ranks==out_rank)high_ranks<-ranks[rank_match:length(ranks)]# Merge read DB and taxid DBreads<-read_db%>%select(-parent_taxid, -rank, -name)%>%left_join(taxid_db, by="taxid")# Extract sequences that are already at appropriate rankreads_rank<-filter(reads, rank==out_rank)# Drop sequences at a higher rank and return unclassified sequencesreads_norank<-reads%>%filter(rank!=out_rank, !rank%in%high_ranks, !is.na(taxid))while(nrow(reads_norank)>0){# As long as there are unclassified sequences...# Promote read taxids and re-merge with taxid DB, then re-classify and filterreads_remaining<-reads_norank%>%mutate(taxid =parent_taxid)%>%select(-parent_taxid, -rank, -name)%>%left_join(taxid_db, by="taxid")reads_rank<-reads_remaining%>%filter(rank==out_rank)%>%bind_rows(reads_rank)reads_norank<-reads_remaining%>%filter(rank!=out_rank, !rank%in%high_ranks, !is.na(taxid))}# Finally, extract and append reads that were excluded during the processreads_dropped<-reads%>%filter(!seq_id%in%reads_rank$seq_id)reads_out<-reads_rank%>%bind_rows(reads_dropped)%>%select(-parent_taxid, -rank, -name)%>%left_join(taxid_db, by="taxid")return(reads_out)}hv_reads_species<-raise_rank(mrg_hv_named, viral_taxa, "species")hv_reads_genus<-raise_rank(mrg_hv_named, viral_taxa, "genus")hv_reads_family<-raise_rank(mrg_hv_named, viral_taxa, "family")
Code
threshold_major_family<-0.06# Count reads for each human-viral familyhv_family_counts<-hv_reads_family%>%group_by(sample, room, sample_type, na_type, name, taxid)%>%count(name ="n_reads_hv")%>%group_by(sample, room, sample_type, na_type)%>%mutate(p_reads_hv =n_reads_hv/sum(n_reads_hv))# Identify high-ranking families and group othershv_family_major_tab<-hv_family_counts%>%group_by(name)%>%filter(p_reads_hv==max(p_reads_hv))%>%filter(row_number()==1)%>%arrange(desc(p_reads_hv))%>%filter(p_reads_hv>threshold_major_family)hv_family_counts_major<-hv_family_counts%>%mutate(name_display =ifelse(name%in%hv_family_major_tab$name, name, "Other"))%>%group_by(sample, room, sample_type, na_type, name_display)%>%summarize(n_reads_hv =sum(n_reads_hv), p_reads_hv =sum(p_reads_hv), .groups="drop")%>%mutate(name_display =factor(name_display, levels =c(hv_family_major_tab$name, "Other")))hv_family_counts_display<-hv_family_counts_major%>%rename(p_reads =p_reads_hv, classification =name_display)# Plotg_hv_family<-g_comp_base+geom_col(data=hv_family_counts_display, position ="stack")+scale_y_continuous(name="% HV Reads", limits=c(0,1.01), breaks =seq(0,1,0.2), expand=c(0,0), labels =function(y)y*100)+scale_fill_manual(values=palette_viral, name ="Viral family")g_hv_family
Code
threshold_major_genus<-0.05# Count reads for each human-viral genushv_genus_counts<-hv_reads_genus%>%filter(na_type=="DNA")%>%group_by(sample, room, sample_type, na_type, name, taxid)%>%count(name ="n_reads_hv")%>%group_by(sample, room, sample_type, na_type)%>%mutate(p_reads_hv =n_reads_hv/sum(n_reads_hv))# Identify high-ranking families and group othershv_genus_major_tab<-hv_genus_counts%>%group_by(name)%>%filter(p_reads_hv==max(p_reads_hv))%>%filter(row_number()==1)%>%arrange(desc(p_reads_hv))%>%filter(p_reads_hv>threshold_major_genus)hv_genus_counts_major<-hv_genus_counts%>%mutate(name_display =ifelse(name%in%hv_genus_major_tab$name, name, "Other"))%>%group_by(sample, room, sample_type, na_type, name_display)%>%summarize(n_reads_hv =sum(n_reads_hv), p_reads_hv =sum(p_reads_hv), .groups="drop")%>%mutate(name_display =factor(name_display, levels =c(hv_genus_major_tab$name, "Other")))hv_genus_counts_display<-hv_genus_counts_major%>%rename(p_reads =p_reads_hv, classification =name_display)# Plotg_hv_genus<-g_comp_base+geom_col(data=hv_genus_counts_display, position ="stack")+scale_y_continuous(name="% HV Reads", limits=c(0,1.01), breaks =seq(0,1,0.2), expand=c(0,0), labels =function(y)y*100)+scale_fill_manual(values=palette_viral, name ="Viral genus")g_hv_genus
Code
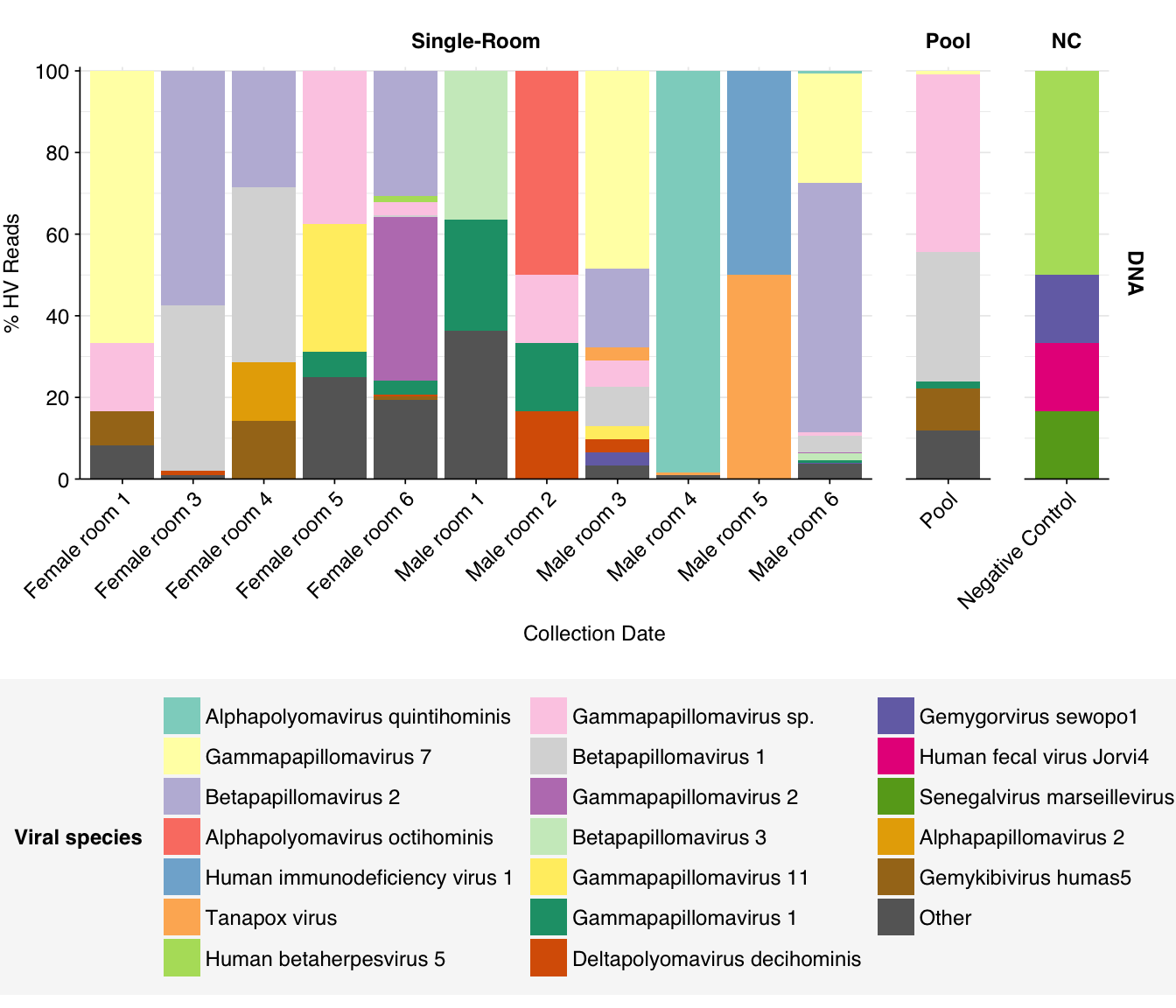
threshold_major_species<-0.1# Count reads for each human-viral specieshv_species_counts<-hv_reads_species%>%filter(na_type=="DNA")%>%group_by(sample, room, sample_type, na_type, name, taxid)%>%count(name ="n_reads_hv")%>%group_by(sample, room, sample_type, na_type)%>%mutate(p_reads_hv =n_reads_hv/sum(n_reads_hv))# Identify high-ranking families and group othershv_species_major_tab<-hv_species_counts%>%group_by(name)%>%filter(p_reads_hv==max(p_reads_hv))%>%filter(row_number()==1)%>%arrange(desc(p_reads_hv))%>%filter(p_reads_hv>threshold_major_species)hv_species_counts_major<-hv_species_counts%>%mutate(name_display =ifelse(name%in%hv_species_major_tab$name, name, "Other"))%>%group_by(sample, room, sample_type, na_type, name_display)%>%summarize(n_reads_hv =sum(n_reads_hv), p_reads_hv =sum(p_reads_hv), .groups="drop")%>%mutate(name_display =factor(name_display, levels =c(hv_species_major_tab$name, "Other")))hv_species_counts_display<-hv_species_counts_major%>%rename(p_reads =p_reads_hv, classification =name_display)# Plotg_hv_species<-g_comp_base+geom_col(data=hv_species_counts_display, position ="stack")+scale_y_continuous(name="% HV Reads", limits=c(0,1.01), breaks =seq(0,1,0.2), expand=c(0,0), labels =function(y)y*100)+scale_fill_manual(values=palette_viral, name ="Viral species")+guides(fill=guide_legend(ncol=3))g_hv_species
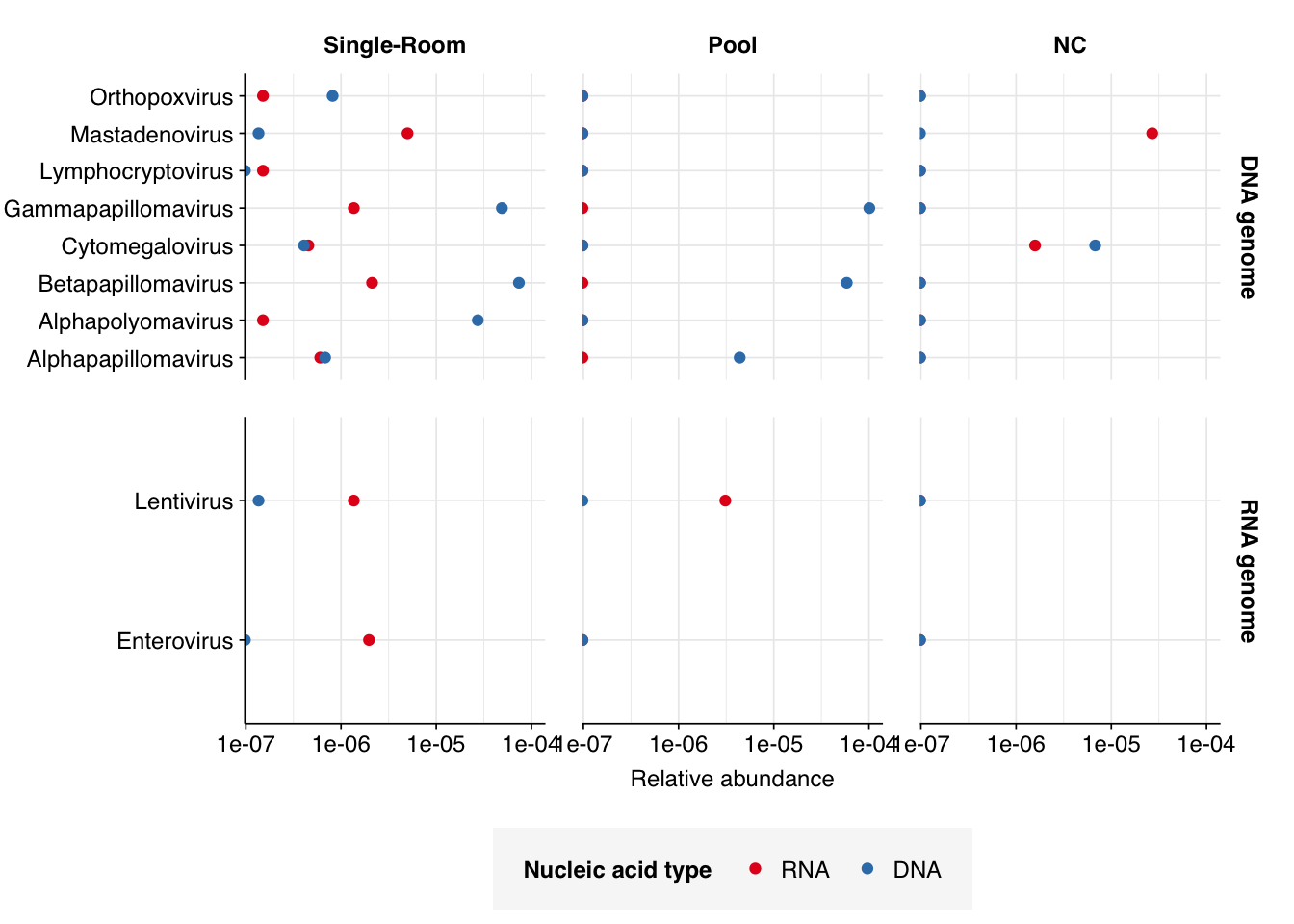
Finally, here again are the overall relative abundances of the specific viral genera I picked out manually in my last entry:
Warning: Transformation introduced infinite values in continuous x-axis
One immediate observation, which I think helps demonstrate the unreliability of the RNA data here, is the almost complete absence of RNA-virus genera of interest. It’s very suspicious for putative HIV reads to be almost as abundant as Enterovirus reads (which, remember, includes rhinovirus), while coronaviruses and influenza are totally absent. The DNA reads seem much more similar to Prussin, though I’m still surprised HCMV and Mastadenovirus are so rare.
Conclusion
This is the second, and by far the smaller, of the air-sampling datasets I’ve analyzed with this pipeline so far. Many of the high-level findings were similar to Prussin, including high relative abundance of human reads, low total viral reads, an absence of enteric viruses, and high abundance of papillomaviruses among human-infecting viruses. The biggest difference in the DNA data was the almost complete lack of cytomegalovirus, compared to its very high abundance in the Prussin data.
Given how small this dataset is (16M reads on a MiSeq, compared to ~450M for Prussin and 1.3B for the upcoming Leung et al. dataset), I don’t think too much weight should be placed on these findings, but given how rare air-MGS datasets are it’s nice to have another one.
Source Code
---title: "Workflow analysis of Rosario et al. (2018)"subtitle: "Air sampling from a student dorm in Colorado."author: "Will Bradshaw"date: 2024-04-12format: html: code-fold: true code-tools: true code-link: true df-print: pagededitor: visualtitle-block-banner: black---```{r}#| label: load-packages#| include: falselibrary(tidyverse)library(cowplot)library(patchwork)library(fastqcr)library(RColorBrewer)source("../scripts/aux_plot-theme.R")theme_base <- theme_base +theme(aspect.ratio =NULL)theme_rotate <- theme_base +theme(axis.text.x =element_text(hjust =1, angle =45),)theme_kit <- theme_rotate +theme(axis.title.x =element_blank(),)tnl <-theme(legend.position ="none")```Continuing our look at air sampling datasets, we turn to [Rosario et al. (2018)](https://pubs.acs.org/doi/10.1021/acs.est.7b04203), another study of air filters, this time from HVAC filters from an undergraduate dorm building at the University of Colorado campus in Boulder. As in [Prussin](https://data.securebio.org/wills-public-notebook/notebooks/2024-04-12_prussin.html), samples were eluted from filters (in this case MERV-8, so less stringent than Prussin's MERV-14 filters) and underwent both RNA and DNA sequencing – this time on an Illumina MiSeq with 2x250bp reads.# The raw dataThe Rosario dataset comprised sequencing data from 12 individual dormitory rooms at the sampling site, as well as a pooled sample of eight additional rooms and a negative control.```{r}#| warning: false#| label: read-qc-data# Data input pathsdata_dir <-"../data/2024-04-12_rosario/"libraries_path <-file.path(data_dir, "sample-metadata.csv")basic_stats_path <-file.path(data_dir, "qc_basic_stats.tsv")adapter_stats_path <-file.path(data_dir, "qc_adapter_stats.tsv")quality_base_stats_path <-file.path(data_dir, "qc_quality_base_stats.tsv")quality_seq_stats_path <-file.path(data_dir, "qc_quality_sequence_stats.tsv")# Import libraries and extract metadata from sample nameslibraries_raw <-read_csv(libraries_path, show_col_types =FALSE)libraries <- libraries_raw %>%arrange(desc(na_type)) %>%mutate(na_type =fct_inorder(na_type)) %>%mutate(sample_type =ifelse(grepl("Control", room), "NC",ifelse(grepl("Pool", room), "Pool", "Single-Room")),sample_type =factor(sample_type, levels=c("Single-Room", "Pool", "NC")),room =ifelse(grepl("Control", room), "Negative Control", room))# Import QC datastages <-c("raw_concat", "cleaned", "dedup", "ribo_initial", "ribo_secondary")basic_stats <-read_tsv(basic_stats_path, show_col_types =FALSE) %>%inner_join(libraries, by="sample") %>%mutate(stage =factor(stage, levels = stages),sample =fct_inorder(sample))adapter_stats <-read_tsv(adapter_stats_path, show_col_types =FALSE) %>%mutate(sample =sub("_2$", "", sample)) %>%inner_join(libraries, by="sample") %>%mutate(stage =factor(stage, levels = stages),read_pair =fct_inorder(as.character(read_pair)))quality_base_stats <-read_tsv(quality_base_stats_path, show_col_types =FALSE) %>%inner_join(libraries, by="sample") %>%mutate(stage =factor(stage, levels = stages),read_pair =fct_inorder(as.character(read_pair)))quality_seq_stats <-read_tsv(quality_seq_stats_path, show_col_types =FALSE) %>%inner_join(libraries, by="sample") %>%mutate(stage =factor(stage, levels = stages),read_pair =fct_inorder(as.character(read_pair)))# Filter to raw databasic_stats_raw <- basic_stats %>%filter(stage =="raw_concat")adapter_stats_raw <- adapter_stats %>%filter(stage =="raw_concat")quality_base_stats_raw <- quality_base_stats %>%filter(stage =="raw_concat")quality_seq_stats_raw <- quality_seq_stats %>%filter(stage =="raw_concat")# Get key values for readoutraw_read_counts <- basic_stats_raw %>%group_by(na_type, sample_type) %>%summarize(rmin =min(n_read_pairs), rmax=max(n_read_pairs),rmean=mean(n_read_pairs), .groups ="drop")raw_read_totals <- basic_stats_raw %>%group_by(na_type, sample_type) %>%summarize(n_read_pairs =sum(n_read_pairs), n_bases_approx =sum(n_bases_approx), .groups ="drop")raw_dup <- basic_stats_raw %>%group_by(na_type, sample_type) %>%summarize(dmin =min(percent_duplicates), dmax=max(percent_duplicates),dmean=mean(percent_duplicates), .groups ="drop")```The 12 single-room samples yielded 0.57-0.74M (mean 0.67M) RNA-sequencing reads and 0.48M-0.84M (mean 0.67M) DNA-sequencing reads; the pooled and control samples had similar depth. In total, non-control samples yielded 8.0M RNA read pairs and 8.0M DNA read pairs (4 gigabases of sequence each).Read qualities were so-so, in need of cleaning. Adapter levels were high. Inferred duplication levels were low (4.6-8.2%) in DNA reads but highly variable (3-83%) in RNA reads.```{r}#| fig-width: 9#| warning: false#| label: plot-basic-stats# Prepare databasic_stats_raw_metrics <- basic_stats_raw %>%select(room, na_type, sample_type,`# Read pairs`= n_read_pairs,`Total base pairs\n(approx)`= n_bases_approx,`% Duplicates\n(FASTQC)`= percent_duplicates) %>%pivot_longer(-(room:sample_type), names_to ="metric", values_to ="value") %>%mutate(metric =fct_inorder(metric))# Set up plot templatesscale_fill_na <- purrr::partial(scale_fill_brewer, palette="Set1", name="Nucleic acid type")g_basic <-ggplot(basic_stats_raw_metrics, aes(x=room, y=value, fill=na_type)) +geom_col(position ="dodge") +scale_x_discrete(name="Collection Date") +scale_y_continuous(expand=c(0,0)) +expand_limits(y=c(0,100)) +scale_fill_na() +facet_grid(metric~sample_type, scales ="free", space="free_x", switch="y") + theme_rotate +theme(axis.title.y =element_blank(),strip.text.y =element_text(face="plain") )g_basic``````{r}#| label: plot-raw-quality# Set up plotting templatesscale_color_na <- purrr::partial(scale_color_brewer, palette="Set1",name="Nucleic acid type")g_qual_raw <-ggplot(mapping=aes(color=na_type, linetype=read_pair, group=interaction(sample,read_pair))) +scale_color_na() +scale_linetype_discrete(name ="Read Pair") +guides(color=guide_legend(nrow=2,byrow=TRUE),linetype =guide_legend(nrow=2,byrow=TRUE)) + theme_base# Visualize adaptersg_adapters_raw <- g_qual_raw +geom_line(aes(x=position, y=pc_adapters), data=adapter_stats_raw) +scale_y_continuous(name="% Adapters", limits=c(0,NA),breaks =seq(0,100,10), expand=c(0,0)) +scale_x_continuous(name="Position", limits=c(0,NA),breaks=seq(0,140,20), expand=c(0,0)) +facet_grid(sample_type~adapter)g_adapters_raw# Visualize qualityg_quality_base_raw <- g_qual_raw +geom_hline(yintercept=25, linetype="dashed", color="red") +geom_hline(yintercept=30, linetype="dashed", color="red") +geom_line(aes(x=position, y=mean_phred_score), data=quality_base_stats_raw) +scale_y_continuous(name="Mean Phred score", expand=c(0,0), limits=c(10,45)) +scale_x_continuous(name="Position", limits=c(0,NA),breaks=seq(0,140,20), expand=c(0,0)) +facet_grid(sample_type~.)g_quality_base_rawg_quality_seq_raw <- g_qual_raw +geom_vline(xintercept=25, linetype="dashed", color="red") +geom_vline(xintercept=30, linetype="dashed", color="red") +geom_line(aes(x=mean_phred_score, y=n_sequences), data=quality_seq_stats_raw) +scale_x_continuous(name="Mean Phred score", expand=c(0,0)) +scale_y_continuous(name="# Sequences", expand=c(0,0)) +facet_grid(sample_type~., scales ="free_y")g_quality_seq_raw```# PreprocessingThe average fraction of reads lost at each stage in the preprocessing pipeline is shown in the following table. Single-room samples lost an average of 52%/14% (RNA/DNA) of total read pairs during cleaning and deduplication, with a further loss of 4.3%/0.1% during ribodepletion. However, the fraction of reads lost, especially during cleaning and deduplication, was highly variable between samples.```{r}#| label: preproc-tablen_reads_rel <- basic_stats %>%select(sample, na_type, sample_type, stage, percent_duplicates, n_read_pairs) %>%group_by(sample, na_type) %>%arrange(sample, na_type, stage) %>%mutate(p_reads_retained =replace_na(n_read_pairs /lag(n_read_pairs), 0),p_reads_lost =1- p_reads_retained,p_reads_retained_abs = n_read_pairs / n_read_pairs[1],p_reads_lost_abs =1-p_reads_retained_abs,p_reads_lost_abs_marginal =replace_na(p_reads_lost_abs -lag(p_reads_lost_abs), 0))n_reads_rel_display <- n_reads_rel %>%rename(Stage=stage, `NA Type`=na_type, `Sample Type`=sample_type) %>%group_by(`Sample Type`, `NA Type`, Stage) %>%summarize(`% Total Reads Lost (Cumulative)`=paste0(round(min(p_reads_lost_abs*100),1), "-", round(max(p_reads_lost_abs*100),1), " (mean ", round(mean(p_reads_lost_abs*100),1), ")"),`% Total Reads Lost (Marginal)`=paste0(round(min(p_reads_lost_abs_marginal*100),1), "-", round(max(p_reads_lost_abs_marginal*100),1), " (mean ", round(mean(p_reads_lost_abs_marginal*100),1), ")"), .groups="drop") %>%filter(Stage !="raw_concat") %>%mutate(Stage = Stage %>% as.numeric %>%factor(labels=c("Trimming & filtering", "Deduplication", "Initial ribodepletion", "Secondary ribodepletion")))n_reads_rel_display``````{r}#| label: preproc-figures#| warning: false#| fig-height: 8#| fig-width: 6g_stage_trace <-ggplot(basic_stats, aes(x=stage, color=na_type, group=sample)) +scale_color_na() +facet_wrap(sample_type~na_type, scales="free", ncol=2,labeller =label_wrap_gen(multi_line=FALSE)) + theme_kit# Plot reads over preprocessingg_reads_stages <- g_stage_trace +geom_line(aes(y=n_read_pairs)) +scale_y_continuous("# Read pairs", expand=c(0,0), limits=c(0,NA))g_reads_stages# Plot relative read losses during preprocessingg_reads_rel <-ggplot(n_reads_rel, aes(x=stage, color=na_type, group=sample)) +geom_line(aes(y=p_reads_lost_abs_marginal)) +scale_y_continuous("% Total Reads Lost", expand=c(0,0), labels =function(x) x*100) +scale_color_na() +facet_wrap(sample_type~na_type, scales="free", ncol=2,labeller =label_wrap_gen(multi_line=FALSE)) + theme_kitg_reads_rel```Data cleaning was very successful at improving read qualities, as well as removing most adapter sequences. The exception to the latter was terminal poly-A sequences, which remained at a substantial (though reduced) prevalence throughout the pipeline. (This doesn't seem to have caused downstream problems that I can see.)```{r}#| warning: false#| label: plot-quality#| fig-height: 7g_qual <-ggplot(mapping=aes(color=na_type, linetype=read_pair, group=interaction(sample,read_pair))) +scale_color_na() +scale_linetype_discrete(name ="Read Pair") +guides(color=guide_legend(nrow=2,byrow=TRUE),linetype =guide_legend(nrow=2,byrow=TRUE)) + theme_base# Visualize adaptersg_adapters <- g_qual +geom_line(aes(x=position, y=pc_adapters), data=adapter_stats) +scale_y_continuous(name="% Adapters", limits=c(0,20),breaks =seq(0,50,10), expand=c(0,0)) +scale_x_continuous(name="Position", limits=c(0,NA),breaks=seq(0,140,20), expand=c(0,0)) +facet_grid(stage~adapter)g_adapters# Visualize qualityg_quality_base <- g_qual +geom_hline(yintercept=25, linetype="dashed", color="red") +geom_hline(yintercept=30, linetype="dashed", color="red") +geom_line(aes(x=position, y=mean_phred_score), data=quality_base_stats) +scale_y_continuous(name="Mean Phred score", expand=c(0,0), limits=c(10,45)) +scale_x_continuous(name="Position", limits=c(0,NA),breaks=seq(0,140,20), expand=c(0,0)) +facet_grid(stage~.)g_quality_baseg_quality_seq <- g_qual +geom_vline(xintercept=25, linetype="dashed", color="red") +geom_vline(xintercept=30, linetype="dashed", color="red") +geom_line(aes(x=mean_phred_score, y=n_sequences), data=quality_seq_stats) +scale_x_continuous(name="Mean Phred score", expand=c(0,0)) +scale_y_continuous(name="# Sequences", expand=c(0,0)) +facet_grid(stage~.)g_quality_seq```According to FASTQC, deduplication was very effective at reducing measured duplicate levels, which for single-room samples fell from an average of 40% to 7.8% for RNA reads and from 5.5% to 0.3% for DNA reads.```{r}#| label: preproc-dedup#| fig-height: 8#| fig-width: 6stage_dup <- basic_stats %>%group_by(na_type, sample_type, stage) %>%summarize(dmin =min(percent_duplicates), dmax=max(percent_duplicates),dmean=mean(percent_duplicates), .groups ="drop")g_dup_stages <- g_stage_trace +geom_line(aes(y=percent_duplicates)) +scale_y_continuous("% Duplicates", limits=c(0,NA), expand=c(0,0))g_dup_stagesg_readlen_stages <- g_stage_trace +geom_line(aes(y=mean_seq_len)) +scale_y_continuous("Mean read length (nt)", expand=c(0,0), limits=c(0,NA))g_readlen_stages```# High-level compositionAs before, to assess the high-level composition of the reads, I ran the ribodepleted files through Kraken (using the Standard 16 database) and summarized the results with Bracken. Combining these results with the read counts above gives us a breakdown of the inferred composition of the samples:```{r}#| label: prepare-composition# Import Bracken databracken_path <-file.path(data_dir, "bracken_counts.tsv")bracken <-read_tsv(bracken_path, show_col_types =FALSE)total_assigned <- bracken %>%group_by(sample) %>%summarize(name ="Total",kraken_assigned_reads =sum(kraken_assigned_reads),added_reads =sum(added_reads),new_est_reads =sum(new_est_reads),fraction_total_reads =sum(fraction_total_reads))bracken_spread <- bracken %>%select(name, sample, new_est_reads) %>%mutate(name =tolower(name)) %>%pivot_wider(id_cols ="sample", names_from ="name", values_from ="new_est_reads")# Count readsread_counts_preproc <- basic_stats %>%select(sample, room, na_type, sample_type, stage, n_read_pairs) %>%pivot_wider(id_cols =c("sample", "room", "na_type", "sample_type"),names_from="stage", values_from="n_read_pairs")read_counts <- read_counts_preproc %>%inner_join(total_assigned %>%select(sample, new_est_reads), by ="sample") %>%rename(assigned = new_est_reads) %>%inner_join(bracken_spread, by="sample")# Assess compositionread_comp <-transmute(read_counts, sample=sample, na_type=na_type,room=room, sample_type=sample_type,n_filtered = raw_concat-cleaned,n_duplicate = cleaned-dedup,n_ribosomal = (dedup-ribo_initial) + (ribo_initial-ribo_secondary),n_unassigned = ribo_secondary-assigned,n_bacterial = bacteria,n_archaeal = archaea,n_viral = viruses,n_human = eukaryota)read_comp_long <-pivot_longer(read_comp, -(sample:sample_type), names_to ="classification",names_prefix ="n_", values_to ="n_reads") %>%mutate(classification =fct_inorder(str_to_sentence(classification))) %>%group_by(sample) %>%mutate(p_reads = n_reads/sum(n_reads))# Summarize compositionread_comp_summ <- read_comp_long %>%group_by(na_type, sample_type, classification) %>%summarize(n_reads =sum(n_reads), .groups ="drop_last") %>%mutate(n_reads =replace_na(n_reads,0),p_reads = n_reads/sum(n_reads),pc_reads = p_reads*100)``````{r}#| label: plot-composition-all# Prepare plotting templatesg_comp_base <-ggplot(mapping=aes(x=room, y=p_reads, fill=classification)) +scale_x_discrete(name="Collection Date") +facet_grid(na_type~sample_type, scales ="free", space ="free_x") + theme_rotatescale_y_pc_reads <- purrr::partial(scale_y_continuous, name ="% Reads",expand =c(0,0), labels =function(y) y*100)# Plot overall compositiong_comp <- g_comp_base +geom_col(data = read_comp_long, position ="stack") +scale_y_pc_reads(limits =c(0,1.01), breaks =seq(0,1,0.2)) +scale_fill_brewer(palette ="Set1", name ="Classification")g_comp# Plot composition of minor componentsread_comp_minor <- read_comp_long %>%filter(classification %in%c("Archaeal", "Viral", "Other"))palette_minor <-brewer.pal(9, "Set1")[c(6,7,9)]g_comp_minor <- g_comp_base +geom_col(data=read_comp_minor, position ="stack") +scale_y_pc_reads() +scale_fill_manual(values=palette_minor, name ="Classification")g_comp_minor``````{r}#| label: composition-summaryp_reads_summ_group <- read_comp_long %>%mutate(classification =ifelse(classification %in%c("Filtered", "Duplicate", "Unassigned"), "Excluded", as.character(classification)),classification =fct_inorder(classification)) %>%group_by(classification, sample, na_type, sample_type) %>%summarize(p_reads =sum(p_reads), .groups ="drop") %>%group_by(classification, na_type, sample_type) %>%summarize(pc_min =min(p_reads)*100, pc_max =max(p_reads)*100, pc_mean =mean(p_reads)*100, .groups ="drop")p_reads_summ_prep <- p_reads_summ_group %>%mutate(classification =fct_inorder(classification),pc_min = pc_min %>%signif(digits=2) %>%sapply(format, scientific=FALSE, trim=TRUE, digits=2),pc_max = pc_max %>%signif(digits=2) %>%sapply(format, scientific=FALSE, trim=TRUE, digits=2),pc_mean = pc_mean %>%signif(digits=2) %>%sapply(format, scientific=FALSE, trim=TRUE, digits=2),display =paste0(pc_min, "-", pc_max, "% (mean ", pc_mean, "%)"))p_reads_summ <- p_reads_summ_prep %>%select(sample_type, classification, na_type, display) %>%pivot_wider(names_from=na_type, values_from = display) %>%arrange(sample_type, classification)p_reads_summ```As with the Prussin data, human reads are very elevated compared to wastewater, making up an average of 21% of read pairs in RNA libraries and 4% for DNA libraries for single-room samples (in both cases, the maximum abundance of human reads across samples is much higher). Viral read abundances are higher than Prussin, averaging 0.08% for single-room RNA libraries and 0.04% for DNA libraries; for some reason, the pooled RNA library shows much higher viral prevalence, at 0.33%. The most obvious difference from the Prussin data is that DNA libraries in Rosario show a much higher fraction of non-ribosomal unassigned reads, with an average of 78% of all reads for single-room libraries.As in Prussin, viral DNA reads were dominated by *Caudoviricetes* phages, with many unclassifiable below the class level. Other prominent viral classes in DNA reads included *Papovaviricetes* (which includes *Polyomaviridae* and *Papillomaviridae*), *Megaviridae* (giant viruses) and *Repensiviricetes* (plant and fungal viruses). Pooled samples also showed a significant level of *Quintoviricetes* (parvoviruses).RNA libraries, meanwhile, were dominated by *Alsuviricetes* (mainly plant viruses) and, notably, *Pisoniviricetes* (which includes coronaviruses, picornaviruses including *Enterovirus*, and caliciviruses including *Norovirus*). *Caudoviricetes*, *Herviviricetes* (herpesviruses) and *Revtraviricetes* (retroviruses + Hep B) were also prominent in some samples.```{r}#| label: extract-viral-taxa# Get viral taxonomyviral_taxa_path <-file.path(data_dir, "viral-taxids.tsv.gz")viral_taxa <-read_tsv(viral_taxa_path, show_col_types =FALSE)# Import Kraken reports & extract viral taxasamples <-as.character(basic_stats_raw$sample)col_names <-c("pc_reads_total", "n_reads_clade", "n_reads_direct","rank", "taxid", "name")report_paths <-paste0(data_dir, "kraken/", samples, ".report.gz")kraken_reports <-lapply(1:length(samples), function(n) read_tsv(report_paths[n], col_names = col_names, show_col_types =FALSE) %>%mutate(sample = samples[n])) %>% bind_rowskraken_reports_viral <-filter(kraken_reports, taxid %in% viral_taxa$taxid) %>%group_by(sample) %>%mutate(p_reads_viral = n_reads_clade/n_reads_clade[1])kraken_reports_viral_cleaned <- kraken_reports_viral %>%inner_join(libraries, by="sample") %>%select(-pc_reads_total, -n_reads_direct) %>%select(name, taxid, p_reads_viral, n_reads_clade, everything())viral_classes <- kraken_reports_viral_cleaned %>%filter(rank =="C")viral_families <- kraken_reports_viral_cleaned %>%filter(rank =="F")``````{r}#| label: viral-class-composition# Identify major viral classesviral_classes_major_tab <- viral_classes %>%group_by(name, taxid) %>%summarize(p_reads_viral_max =max(p_reads_viral), .groups="drop") %>%filter(p_reads_viral_max >=0.04)viral_classes_major_list <- viral_classes_major_tab %>%pull(name)viral_classes_major <- viral_classes %>%filter(name %in% viral_classes_major_list) %>%select(name, taxid, sample, room, na_type, sample_type, p_reads_viral)viral_classes_minor <- viral_classes_major %>%group_by(sample, room, na_type, sample_type) %>%summarize(p_reads_viral_major =sum(p_reads_viral), .groups ="drop") %>%mutate(name ="Other", taxid=NA, p_reads_viral =1-p_reads_viral_major) %>%select(name, taxid, sample, room, na_type, sample_type, p_reads_viral)viral_classes_display <-bind_rows(viral_classes_major, viral_classes_minor) %>%arrange(desc(p_reads_viral)) %>%mutate(name =factor(name, levels=c(viral_classes_major_list, "Other")),p_reads_viral =pmax(p_reads_viral, 0)) %>%rename(p_reads = p_reads_viral, classification=name)palette_viral <-c(brewer.pal(12, "Set3"), brewer.pal(8, "Dark2"))g_classes <- g_comp_base +geom_col(data=viral_classes_display, position ="stack") +scale_y_continuous(name="% Viral Reads", limits=c(0,1.01), breaks =seq(0,1,0.2),expand=c(0,0), labels =function(y) y*100) +scale_fill_manual(values=palette_viral, name ="Viral class")g_classes```Of these, the strong presence of *Papovaviricetes* and *Pisoniviricetes* across multiple samples is the most interesting, as both contain important human pathogens. The presence of *Quintoviricetes*, *Herviviricetes* and *Revtraviricetes* in a smaller number of samples is also notable.In DNA reads, *Papovaviricetes* are quite heterogeneous across samples, while mostly being dominated by one or two genera within samples. The three rooms with the largest *Papovaviricetes* fraction (Female room 1 & 3 and Male room 4) are each dominated by a single genus, specifically *Dyothetapapillomavirus* (of which the sole member is *Dyothetapapillomavirus 1*, a cat pathogen), *Betapapillomavirus* (a genus of human wart-causing viruses), and *Alphapolyomavirus* (specifically Merkel cell polyomavirus).```{r}#| label: papovaviricetes# Get all read counts in classpapova_taxid <-2732421papova_desc_taxids_old <- papova_taxidpapova_desc_taxids_new <-unique(c(papova_desc_taxids_old, viral_taxa %>%filter(parent_taxid %in% papova_desc_taxids_old) %>%pull(taxid)))while (length(papova_desc_taxids_new) >length(papova_desc_taxids_old)){ papova_desc_taxids_old <- papova_desc_taxids_new papova_desc_taxids_new <-unique(c(papova_desc_taxids_old, viral_taxa %>%filter(parent_taxid %in% papova_desc_taxids_old) %>%pull(taxid)))}papova_counts <- kraken_reports_viral_cleaned %>%filter(taxid %in% papova_desc_taxids_new) %>%mutate(p_reads_papova = n_reads_clade/n_reads_clade[1])# Get genus compositionpapova_genera <- papova_counts %>%filter(rank =="G", na_type =="DNA")papova_genera_major_tab <- papova_genera %>%group_by(name, taxid) %>%summarize(p_reads_papova_max =max(p_reads_papova), .groups="drop") %>%filter(p_reads_papova_max >=0.04)papova_genera_major_list <- papova_genera_major_tab %>%pull(name)papova_genera_major <- papova_genera %>%filter(name %in% papova_genera_major_list) %>%select(name, taxid, sample, room, na_type, sample_type, p_reads_papova)papova_genera_minor <- papova_genera_major %>%group_by(sample, room, na_type, sample_type) %>%summarize(p_reads_papova_major =sum(p_reads_papova), .groups ="drop") %>%mutate(name ="Other", taxid=NA, p_reads_papova =1-p_reads_papova_major) %>%select(name, taxid, sample, room, na_type, sample_type, p_reads_papova)papova_genera_display <-bind_rows(papova_genera_major, papova_genera_minor) %>%arrange(desc(p_reads_papova)) %>%mutate(name =factor(name, levels=c(papova_genera_major_list, "Other"))) %>%rename(p_reads = p_reads_papova, classification=name)# Plotg_papova_genera <- g_comp_base +geom_col(data=papova_genera_display, position ="stack") +scale_y_continuous(name="% Papovaviricetes Reads", limits=c(0,1.01), breaks =seq(0,1,0.2),expand=c(0,0), labels =function(y) y*100) +scale_fill_manual(values=palette_viral, name ="Viral genus") +guides(fill=guide_legend(ncol=3))g_papova_genera```In RNA reads, *Pisoniviricetes* are conversely dominated by a single genus, *Sobemovirus*, across all samples, and specifically a single species. Disappointingly, this is Ryegrass mottle virus, a plant pathogen.```{r}#| label: pisoniviricetes# Get all read counts in classpisoni_taxid <-2732506pisoni_desc_taxids_old <- pisoni_taxidpisoni_desc_taxids_new <-unique(c(pisoni_desc_taxids_old, viral_taxa %>%filter(parent_taxid %in% pisoni_desc_taxids_old) %>%pull(taxid)))while (length(pisoni_desc_taxids_new) >length(pisoni_desc_taxids_old)){ pisoni_desc_taxids_old <- pisoni_desc_taxids_new pisoni_desc_taxids_new <-unique(c(pisoni_desc_taxids_old, viral_taxa %>%filter(parent_taxid %in% pisoni_desc_taxids_old) %>%pull(taxid)))}pisoni_counts <- kraken_reports_viral_cleaned %>%filter(taxid %in% pisoni_desc_taxids_new) %>%mutate(p_reads_pisoni = n_reads_clade/n_reads_clade[1])# Get genus compositionpisoni_genera <- pisoni_counts %>%filter(rank =="S", na_type =="RNA")pisoni_genera_major_tab <- pisoni_genera %>%group_by(name, taxid) %>%summarize(p_reads_pisoni_max =max(p_reads_pisoni), .groups="drop") %>%filter(p_reads_pisoni_max >=0.04)pisoni_genera_major_list <- pisoni_genera_major_tab %>%pull(name)pisoni_genera_major <- pisoni_genera %>%filter(name %in% pisoni_genera_major_list) %>%select(name, taxid, sample, room, na_type, sample_type, p_reads_pisoni)pisoni_genera_minor <- pisoni_genera_major %>%group_by(sample, room, na_type, sample_type) %>%summarize(p_reads_pisoni_major =sum(p_reads_pisoni), .groups ="drop") %>%mutate(name ="Other", taxid=NA, p_reads_pisoni =1-p_reads_pisoni_major) %>%select(name, taxid, sample, room, na_type, sample_type, p_reads_pisoni)pisoni_genera_display <-bind_rows(pisoni_genera_major, pisoni_genera_minor) %>%arrange(desc(p_reads_pisoni)) %>%mutate(name =factor(name, levels=c(pisoni_genera_major_list, "Other"))) %>%rename(p_reads = p_reads_pisoni, classification=name)# Plotg_pisoni_genera <- g_comp_base +geom_col(data=pisoni_genera_display, position ="stack") +scale_y_continuous(name="% Pisoniviricetes Reads", limits=c(0,1.01), breaks =seq(0,1,0.2),expand=c(0,0), labels =function(y) y*100) +scale_fill_manual(values=palette_viral, name ="Viral genus") +guides(fill=guide_legend(ncol=3))g_pisoni_genera```# Human-infecting virus reads: validationNext, I investigated the human-infecting virus read content of these unenriched samples. Using the same workflow I used for Prussin et al, I identified 118 RNA read pairs and 1269 DNA read pairs as putatively human viral: 0.004% and 0.017% of surviving reads, respectively.```{r}#| label: hv-read-countshv_reads_filtered_path <-file.path(data_dir, "hv_hits_putative_filtered.tsv.gz")hv_reads_filtered <-read_tsv(hv_reads_filtered_path, show_col_types =FALSE) %>%inner_join(libraries, by="sample") %>%arrange(na_type, sample_type)n_hv_filtered <- hv_reads_filtered %>%group_by(sample, room, na_type, sample_type) %>% count %>%inner_join(basic_stats %>%filter(stage =="ribo_initial") %>%select(sample, n_read_pairs), by="sample") %>%rename(n_putative = n, n_total = n_read_pairs) %>%mutate(p_reads = n_putative/n_total, pc_reads = p_reads *100)n_hv_filtered_summ <- n_hv_filtered %>%group_by(na_type) %>%summarize(n_putative =sum(n_putative), n_total =sum(n_total), .groups="drop") %>%mutate(p_reads = n_putative/n_total, pc_reads = p_reads*100)``````{r}#| label: plot-hv-scores#| warning: false#| fig-width: 8mrg <- hv_reads_filtered %>%mutate(kraken_label =ifelse(assigned_hv, "Kraken2 HV\nassignment",ifelse(hit_hv, "Kraken2 HV\nhit","No hit or\nassignment"))) %>%group_by(sample, na_type) %>%arrange(desc(adj_score_fwd), desc(adj_score_rev)) %>%mutate(seq_num =row_number(),adj_score_max =pmax(adj_score_fwd, adj_score_rev))# Import Bowtie2/Kraken data and perform filtering stepsg_mrg <-ggplot(mrg, aes(x=adj_score_fwd, y=adj_score_rev)) +geom_point(alpha=0.5, shape=16) +scale_x_continuous(name="S(forward read)", limits=c(0,65), breaks=seq(0,100,10), expand =c(0,0)) +scale_y_continuous(name="S(reverse read)", limits=c(0,65), breaks=seq(0,100,10), expand =c(0,0)) +facet_grid(na_type~kraken_label, labeller =labeller(kit =label_wrap_gen(20))) + theme_base +theme(aspect.ratio=1)g_mrgg_hist_0 <-ggplot(mrg, aes(x=adj_score_max)) +geom_histogram(binwidth=5,boundary=0,position="dodge") +facet_grid(na_type~kraken_label, scales="free_y") +scale_x_continuous(name ="Maximum adjusted alignment score") +scale_y_continuous(name="# Read pairs") +scale_fill_brewer(palette ="Dark2") + theme_base +geom_vline(xintercept=20, linetype="dashed", color="red")g_hist_0```As previously described, I ran BLASTN on these reads via a dedicated EC2 instance, using the same parameters I've used for previous datasets.```{r}#| label: make-blast-fastamrg_fasta <- mrg %>%mutate(seq_head =paste0(">", seq_id)) %>% ungroup %>%select(header1=seq_head, seq1=query_seq_fwd, header2=seq_head, seq2=query_seq_rev) %>%mutate(header1=paste0(header1, "_1"), header2=paste0(header2, "_2"))mrg_fasta_out <-do.call(paste, c(mrg_fasta, sep="\n")) %>%paste(collapse="\n")write(mrg_fasta_out, file.path(data_dir, "blast/putative-viral.fasta"))``````{r}#| label: process-blast-data#| warning: false# Import BLAST resultsblast_results_path <-file.path(data_dir, "blast/putative-viral.blast.gz")blast_cols <-c("qseqid", "sseqid", "sgi", "staxid", "qlen", "evalue", "bitscore", "qcovs", "length", "pident", "mismatch", "gapopen", "sstrand", "qstart", "qend", "sstart", "send")blast_results <-read_tsv(blast_results_path, show_col_types =FALSE,col_names = blast_cols, col_types =cols(.default="c"))# blast_results_path <- file.path(data_dir, "blast/putative-viral-best.blast.gz")# blast_results <- read_tsv(blast_results_path, show_col_types = FALSE)# Filter for best hit for each query/subject combinationblast_results_best <- blast_results %>%group_by(qseqid, staxid) %>%filter(bitscore ==max(bitscore)) %>%filter(length ==max(length)) %>%filter(row_number() ==1)# Rank hits for each query and filter for high-ranking hitsblast_results_ranked <- blast_results_best %>%group_by(qseqid) %>%mutate(rank =dense_rank(desc(bitscore)))blast_results_highrank <- blast_results_ranked %>%filter(rank <=5) %>%mutate(read_pair =str_split(qseqid, "_") %>%sapply(nth, n=-1), seq_id =str_split(qseqid, "_") %>%sapply(nth, n=1)) %>%mutate(bitscore =as.numeric(bitscore))# Summarize by read pair and taxidblast_results_paired <- blast_results_highrank %>%group_by(seq_id, staxid) %>%summarize(bitscore_max =max(bitscore), bitscore_min =min(bitscore),n_reads =n(), .groups ="drop")# Add viral statusblast_results_viral <-mutate(blast_results_paired, viral = staxid %in% viral_taxa$taxid) %>%mutate(viral_full = viral & n_reads ==2)# Compare to Kraken & Bowtie assignmentsmrg_assign <- mrg %>%select(sample, seq_id, taxid, assigned_taxid, adj_score_max, na_type)blast_results_assign <-left_join(blast_results_viral, mrg_assign, by="seq_id") %>%mutate(taxid_match_bowtie = (staxid == taxid),taxid_match_kraken = (staxid == assigned_taxid),taxid_match_any = taxid_match_bowtie | taxid_match_kraken)blast_results_out <- blast_results_assign %>%group_by(seq_id) %>%summarize(viral_status =ifelse(any(viral_full), 2,ifelse(any(taxid_match_any), 2,ifelse(any(viral), 1, 0))),.groups ="drop")``````{r}#| label: plot-blast-results#| fig-height: 6#| warning: false# Merge BLAST results with unenriched read datamrg_blast <-full_join(mrg, blast_results_out, by="seq_id") %>%mutate(viral_status =replace_na(viral_status, 0),viral_status_out =ifelse(viral_status ==0, FALSE, TRUE))mrg_blast_rna <- mrg_blast %>%filter(na_type =="RNA")mrg_blast_dna <- mrg_blast %>%filter(na_type =="DNA")# Plot RNAg_mrg_blast_rna <- mrg_blast_rna %>%ggplot(aes(x=adj_score_fwd, y=adj_score_rev, color=viral_status_out)) +geom_point(alpha=0.5, shape=16) +scale_x_continuous(name="S(forward read)", limits=c(0,65), breaks=seq(0,100,10), expand =c(0,0)) +scale_y_continuous(name="S(reverse read)", limits=c(0,65), breaks=seq(0,100,10), expand =c(0,0)) +scale_color_brewer(palette ="Set1", name ="Viral status") +facet_grid(viral_status_out~kraken_label, labeller =labeller(kit =label_wrap_gen(20))) + theme_base +labs(title="RNA") +theme(aspect.ratio=1, plot.title =element_text(size=rel(2), hjust=0))g_mrg_blast_rna# Plot DNAg_mrg_blast_dna <- mrg_blast_dna %>%ggplot(aes(x=adj_score_fwd, y=adj_score_rev, color=viral_status_out)) +geom_point(alpha=0.5, shape=16) +scale_x_continuous(name="S(forward read)", limits=c(0,65), breaks=seq(0,100,10), expand =c(0,0)) +scale_y_continuous(name="S(reverse read)", limits=c(0,65), breaks=seq(0,100,10), expand =c(0,0)) +scale_color_brewer(palette ="Set1", name ="Viral status") +facet_grid(viral_status_out~kraken_label, labeller =labeller(kit =label_wrap_gen(20))) + theme_base +labs(title="DNA") +theme(aspect.ratio=1, plot.title =element_text(size=rel(2), hjust=0))g_mrg_blast_dna``````{r}#| label: plot-blast-histogramg_hist_1 <-ggplot(mrg_blast, aes(x=adj_score_max)) +geom_histogram(binwidth=5,boundary=0,position="dodge") +facet_grid(na_type~viral_status_out, scales ="free_y") +scale_x_continuous(name ="Maximum adjusted alignment score") +scale_y_continuous(name="# Read pairs") +scale_fill_brewer(palette ="Dark2") + theme_base +geom_vline(xintercept=20, linetype="dashed", color="red")g_hist_1```These results look good on visual inspection, and indeed precision and sensitivity are both very high. For a disjunctive score threshold of 20, my updated workflow achieves an F1 score of 98.2% for RNA sequences and 97.9% for DNA sequences.```{r}#| label: plot-f1test_sens_spec <-function(tab, score_threshold, conjunctive, include_special){if (!include_special) tab <-filter(tab, viral_status_out %in%c("TRUE", "FALSE")) tab_retained <- tab %>%mutate(conjunctive = conjunctive,retain_score_conjunctive = (adj_score_fwd > score_threshold & adj_score_rev > score_threshold), retain_score_disjunctive = (adj_score_fwd > score_threshold | adj_score_rev > score_threshold),retain_score =ifelse(conjunctive, retain_score_conjunctive, retain_score_disjunctive),retain = assigned_hv | hit_hv | retain_score) %>%group_by(viral_status_out, retain) %>% count pos_tru <- tab_retained %>%filter(viral_status_out =="TRUE", retain) %>%pull(n) %>% sum pos_fls <- tab_retained %>%filter(viral_status_out !="TRUE", retain) %>%pull(n) %>% sum neg_tru <- tab_retained %>%filter(viral_status_out !="TRUE", !retain) %>%pull(n) %>% sum neg_fls <- tab_retained %>%filter(viral_status_out =="TRUE", !retain) %>%pull(n) %>% sum sensitivity <- pos_tru / (pos_tru + neg_fls) specificity <- neg_tru / (neg_tru + pos_fls) precision <- pos_tru / (pos_tru + pos_fls) f1 <-2* precision * sensitivity / (precision + sensitivity) out <-tibble(threshold=score_threshold, include_special = include_special, conjunctive = conjunctive, sensitivity=sensitivity, specificity=specificity, precision=precision, f1=f1)return(out)}range_f1 <-function(intab, inc_special, inrange=15:45){ tss <- purrr::partial(test_sens_spec, tab=intab, include_special=inc_special) stats_conj <-lapply(inrange, tss, conjunctive=TRUE) %>% bind_rows stats_disj <-lapply(inrange, tss, conjunctive=FALSE) %>% bind_rows stats_all <-bind_rows(stats_conj, stats_disj) %>%pivot_longer(!(threshold:conjunctive), names_to="metric", values_to="value") %>%mutate(conj_label =ifelse(conjunctive, "Conjunctive", "Disjunctive"))return(stats_all)}inc_special <-FALSEstats_rna <-range_f1(mrg_blast_rna, inc_special) %>%mutate(na_type ="RNA")stats_dna <-range_f1(mrg_blast_dna, inc_special) %>%mutate(na_type ="DNA")stats_0 <-bind_rows(stats_rna, stats_dna) %>%mutate(attempt=0)threshold_opt_0 <- stats_0 %>%group_by(conj_label,attempt,na_type) %>%filter(metric =="f1") %>%filter(value ==max(value)) %>%filter(threshold ==min(threshold))g_stats_0 <-ggplot(stats_0, aes(x=threshold, y=value, color=metric)) +geom_vline(data = threshold_opt_0, mapping =aes(xintercept=threshold),color ="red", linetype ="dashed") +geom_line() +scale_y_continuous(name ="Value", limits=c(0,1), breaks =seq(0,1,0.2), expand =c(0,0)) +scale_x_continuous(name ="Threshold", expand =c(0,0)) +scale_color_brewer(palette="Set3") +facet_grid(na_type~conj_label) + theme_baseg_stats_0```Looking into the composition of different read groups, the most notable observations for me are the predominance of (1) HIV reads among low-scoring RNA "true-positives", and (2) human mastadenovirus C among RNA true-positives in general:```{r}#| label: fp#| fig-height: 6viral_taxa$name[viral_taxa$taxid ==211787] <-"Human papillomavirus type 92"viral_taxa$name[viral_taxa$taxid ==509154] <-"Porcine endogenous retrovirus C"viral_taxa$name[viral_taxa$taxid ==493803] <-"Merkel cell polyomavirus"viral_taxa$name[viral_taxa$taxid ==427343] <-"Human papillomavirus 107"major_threshold <-0.1fp <- mrg_blast %>%group_by(na_type, viral_status_out, highscore = adj_score_max >=20, taxid) %>% count %>%group_by(na_type, viral_status_out, highscore) %>%mutate(p=n/sum(n)) %>%left_join(viral_taxa, by="taxid") %>%arrange(desc(p)) %>%mutate(name =ifelse(taxid ==194958, "Porcine endogenous retrovirus A", name))fp_major_tab <- fp %>%filter(p > major_threshold) %>%arrange(desc(p))fp_major_list <- fp_major_tab %>%pull(name) %>% sort %>% unique %>%c(., "Other")fp_major <- fp %>%mutate(major = p > major_threshold) %>%mutate(name_display =ifelse(major, name, "Other")) %>%group_by(na_type, viral_status_out, highscore, name_display) %>%summarize(n=sum(n), p=sum(p), .groups ="drop") %>%mutate(name_display =factor(name_display, levels = fp_major_list),score_display =ifelse(highscore, "S >= 20", "S < 20"),status_display =ifelse(viral_status_out, "True positive", "False positive"))g_fp <-ggplot(fp_major, aes(x=score_display, y=p, fill=name_display)) +geom_col(position="stack") +scale_x_discrete(name ="True positive?") +scale_y_continuous(name ="% reads", limits =c(0,1.01), breaks =seq(0,1,0.2), expand =c(0,0)) +scale_fill_manual(values = palette_viral, name ="Viral\ntaxon") +facet_grid(na_type~status_display) +guides(fill=guide_legend(ncol=3)) + theme_kitg_fp```There are only eight low-scoring true-positive RNA reads, three of which are HIV. These do seem to be real; at least, they don't BLAST to anything else. Ironically the eight high-scoring "true-positive" HIV reads seem more likely to be fake; they also map to a range of cloning vectors:```{r}#| label: hiv-blast-hits# Configureref_taxid_hiv <-11676p_threshold <-0.3# Get taxon namestax_names_path <-file.path(data_dir, "taxid-names.tsv.gz")tax_names <-read_tsv(tax_names_path, show_col_types =FALSE)# Add missing namestax_names_new <-tribble(~staxid, ~name,3050295, "Cytomegalovirus humanbeta5",459231, "FLAG-tagging vector pFLAG97-TSR")tax_names <-bind_rows(tax_names, tax_names_new)ref_name_hiv <- tax_names %>%filter(staxid == ref_taxid_hiv) %>%pull(name)# Get major matchesfp_staxid <- mrg_blast %>%filter(taxid == ref_taxid_hiv) %>%group_by(na_type, highscore = adj_score_max >=20) %>%mutate(n_seq =n()) %>%left_join(blast_results_paired, by="seq_id") %>%mutate(staxid =as.integer(staxid)) %>%left_join(tax_names, by="staxid") %>%rename(sname=name) %>%left_join(tax_names %>%rename(taxid=staxid), by="taxid")fp_staxid_count <- fp_staxid %>%group_by(viral_status_out, highscore, na_type, taxid, name, staxid, sname, n_seq) %>% count %>%group_by(viral_status_out, highscore, na_type, taxid, name) %>%mutate(p=n/n_seq)fp_staxid_count_major <- fp_staxid_count %>%filter(n>1, p>p_threshold, !is.na(staxid)) %>%mutate(score_display =ifelse(highscore, "S >= 20", "S < 20"),status_display =ifelse(viral_status_out, "True positive", "False positive"))# Plotg <-ggplot(fp_staxid_count_major, aes(x=p, y=sname)) +geom_col() +facet_grid(na_type~status_display+score_display, scales="free",labeller =label_wrap_gen(multi_line =FALSE)) +scale_x_continuous(name="% mapped reads", limits=c(0,1), breaks=seq(0,1,0.2),expand=c(0,0)) +labs(title=paste0(ref_name_hiv, " (taxid ", ref_taxid_hiv, ")")) + theme_base +theme(axis.title.y =element_blank(),plot.title =element_text(size=rel(1.4), hjust=0, face="plain"))g```A similar pattern is seen for the Human mastadenovirus C hits: low-scoring true-positives only map to adenoviruses, but high-scoring ones also map to a range of cloning vectors.```{r}#| label: masta-blast-hits# Configureref_taxid_masta <-129951p_threshold <-0.1# Add missing namestax_names_new <-tribble(~staxid, ~name,3050295, "Cytomegalovirus humanbeta5",459231, "FLAG-tagging vector pFLAG97-TSR",3033760, "Human adenovirus 89",3043599, "Human adenovirus 108",3088344, "Human adenovirus C108",3102992, "Cloning vector pBWH-C5",3102994, "Cloning vector pBWH-C5-pIX-Kan",3102995, "Cloning vector pBWH-C5-pIXZG" )tax_names <-bind_rows(tax_names, tax_names_new)ref_name_masta <- tax_names %>%filter(staxid == ref_taxid_masta) %>%pull(name)# Get major matchesfp_staxid <- mrg_blast %>%filter(taxid == ref_taxid_masta) %>%group_by(na_type, highscore = adj_score_max >=20) %>%mutate(n_seq =n()) %>%left_join(blast_results_paired, by="seq_id") %>%mutate(staxid =as.integer(staxid)) %>%left_join(tax_names, by="staxid") %>%rename(sname=name) %>%left_join(tax_names %>%rename(taxid=staxid), by="taxid")fp_staxid_count <- fp_staxid %>%group_by(viral_status_out, highscore, na_type, taxid, name, staxid, sname, n_seq) %>% count %>%group_by(viral_status_out, highscore, na_type, taxid, name) %>%mutate(p=n/n_seq)fp_staxid_count_major <- fp_staxid_count %>%filter(p>p_threshold, !is.na(staxid)) %>%mutate(score_display =ifelse(highscore, "S >= 20", "S < 20"),status_display =ifelse(viral_status_out, "True positive", "False positive"))# Plotg <-ggplot(fp_staxid_count_major, aes(x=p, y=sname)) +geom_col() +facet_grid(na_type~status_display+score_display, scales="free",labeller =label_wrap_gen(multi_line =FALSE)) +scale_x_continuous(name="% mapped reads", limits=c(0,1), breaks=seq(0,1,0.2),expand=c(0,0)) +labs(title=paste0(ref_name_masta, " (taxid ", ref_taxid_masta, ")")) + theme_base +theme(axis.title.y =element_blank(),plot.title =element_text(size=rel(1.5), hjust=0, face="plain"))g```The plots above only tell us the total number of reads that BLAST to each cloning vector; they don't tell us how many reads map to any vector. In particular, they can't distinguish between a situation where many reads map to a few cloning vectors, and one where a few reads map to many. This turns out to be about 35% of high-scoring HIV RNA reads and 55% of high-scoring mastadenovirus RNA reads:```{r}#| label: hiv-vector-hitsfp_hiv_vec <- mrg_blast %>%filter(taxid == ref_taxid_hiv) %>%left_join(blast_results_paired, by="seq_id") %>%mutate(staxid =as.integer(staxid)) %>%left_join(tax_names, by="staxid") %>%rename(sname=name) %>%left_join(tax_names %>%rename(taxid=staxid), by="taxid") %>%group_by(vector =grepl("vector", sname))fp_hiv_vec_match <- fp_hiv_vec %>%group_by(viral_status_out, seq_id, na_type, highscore = adj_score_max >=20) %>%summarize(vector_match =any(vector), .groups ="drop")fp_hiv_vec_count <- fp_hiv_vec_match %>%group_by(viral_status_out, na_type, highscore, vector_match) %>% count %>%group_by(viral_status_out, na_type, highscore) %>%mutate(p=n/sum(n)) %>%mutate(score_display =ifelse(highscore, "S >= 20", "S < 20"),status_display =ifelse(viral_status_out,"True positive", "False positive"))g_hiv_vec_count <-ggplot(fp_hiv_vec_count, aes(y=score_display, x=p, fill=vector_match)) +geom_col() +scale_x_continuous(name="% Mapped Reads", limits=c(0,1), breaks=seq(0,1,0.2),expand=c(0,0)) +scale_fill_brewer(name="Maps to vector?", palette ="Dark2") +facet_grid(na_type~status_display, scales="free") +labs(title=paste0(ref_name_hiv, " (taxid ", ref_taxid_hiv, ")")) + theme_base +theme(axis.title.y =element_blank(),plot.title =element_text(size=rel(1.4), hjust=0, face="plain"))g_hiv_vec_count``````{r}#| label: masta-vector-hitsfp_masta_vec <- mrg_blast %>%filter(taxid == ref_taxid_masta) %>%left_join(blast_results_paired, by="seq_id") %>%mutate(staxid =as.integer(staxid)) %>%left_join(tax_names, by="staxid") %>%rename(sname=name) %>%left_join(tax_names %>%rename(taxid=staxid), by="taxid") %>%group_by(vector =grepl("vector", sname))fp_masta_vec_match <- fp_masta_vec %>%group_by(viral_status_out, seq_id, na_type, highscore = adj_score_max >=20) %>%summarize(vector_match =any(vector), .groups ="drop")fp_masta_vec_count <- fp_masta_vec_match %>%group_by(viral_status_out, na_type, highscore, vector_match) %>% count %>%group_by(viral_status_out, na_type, highscore) %>%mutate(p=n/sum(n)) %>%mutate(score_display =ifelse(highscore, "S >= 20", "S < 20"),status_display =ifelse(viral_status_out,"True positive", "False positive"))g_masta_vec_count <-ggplot(fp_masta_vec_count, aes(y=score_display, x=p, fill=vector_match)) +geom_col() +scale_x_continuous(name="% Mapped Reads", limits=c(0,1), breaks=seq(0,1,0.2),expand=c(0,0)) +scale_fill_brewer(name="Maps to vector?", palette ="Dark2") +facet_grid(na_type~status_display, scales="free") +labs(title=paste0(ref_name_masta, " (taxid ", ref_taxid_masta, ")")) + theme_base +theme(axis.title.y =element_blank(),plot.title =element_text(size=rel(1.4), hjust=0, face="plain"))g_masta_vec_count```This is more concerning for the mastadenovirus reads, which make up a nontrivial fraction of all high-scoring RNA "true-positives". As in my last post, though, it's hard to know what to take from this; many vectors are based on lentiviral or adenoviral backbones, and it's not always easy to distinguish between true virus and virus-based vector. I'm going to stick with my pipeline as it is for now, but it's worth keeping this in mind in interpreting the results.# Human-infecting viruses: overall relative abundance```{r}#| label: count-hv-reads# Get raw read countsread_counts_raw <- basic_stats_raw %>%select(sample, room, na_type, sample_type, n_reads_raw = n_read_pairs)# Get HV read countsmrg_hv <- mrg %>%mutate(hv_status = assigned_hv | hit_hv | adj_score_max >=20)read_counts_hv <- mrg_hv %>%filter(hv_status) %>%group_by(sample) %>%count(name="n_reads_hv")read_counts <- read_counts_raw %>%left_join(read_counts_hv, by="sample") %>%mutate(n_reads_hv =replace_na(n_reads_hv, 0))# Aggregateread_counts_total <- read_counts %>%group_by(na_type, sample_type) %>%filter(n() >1) %>%summarize(n_reads_raw =sum(n_reads_raw),n_reads_hv =sum(n_reads_hv), .groups="drop") %>%mutate(sample="All samples", room ="All rooms")read_counts_agg <- read_counts %>%arrange(sample) %>%bind_rows(read_counts_total) %>%mutate(sample =fct_inorder(sample),p_reads_hv = n_reads_hv/n_reads_raw)```In non-control samples, applying a disjunctive cutoff at S=20 identifies 97 RNA reads and 1204 DNA reads as human-viral. This gives an overall relative HV abundance of $1.21 \times 10^{-5}$ for RNA reads and $1.50 \times 10^{-4}$ for DNA reads. For DNA reads, HV RA in the negative controls is about 10x lower than the non-control samples; however, for RNA reads, the HV RA in the negative controls is actually higher than in the non-controls. This, combined with the very low absolute read count, suggests that we shouldn't take the HV results for RNA reads too seriously.```{r}#| label: plot-hv-ra#| warning: false# Visualizeg_phv_agg <-ggplot(read_counts_agg, aes(x=room, color=na_type)) +geom_point(aes(y=p_reads_hv)) +scale_y_log10("Relative abundance of human virus reads") +scale_x_discrete(name="Collection Date") +facet_grid(.~sample_type, scales ="free", space ="free_x") +scale_color_na() + theme_rotateg_phv_agg``````{r}#| label: ra-hv-past# Collate past RA valuesra_past <-tribble(~dataset, ~ra, ~na_type, ~panel_enriched,"Brumfield", 5e-5, "RNA", FALSE,"Brumfield", 3.66e-7, "DNA", FALSE,"Spurbeck", 5.44e-6, "RNA", FALSE,"Yang", 3.62e-4, "RNA", FALSE,"Rothman (unenriched)", 1.87e-5, "RNA", FALSE,"Rothman (panel-enriched)", 3.3e-5, "RNA", TRUE,"Crits-Christoph (unenriched)", 1.37e-5, "RNA", FALSE,"Crits-Christoph (panel-enriched)", 1.26e-2, "RNA", TRUE,"Prussin (non-control)", 1.63e-5, "RNA", FALSE,"Prussin (non-control)", 4.16e-5, "DNA", FALSE)# Collate new RA valuesra_new <-tribble(~dataset, ~ra, ~na_type, ~panel_enriched,"Rosario (non-control)", 1.21e-5, "RNA", FALSE,"Rosario (non-control)", 1.50e-4, "DNA", FALSE)# Plotra_comp <-bind_rows(ra_past, ra_new) %>%mutate(dataset =fct_inorder(dataset))g_ra_comp <-ggplot(ra_comp, aes(y=dataset, x=ra, color=na_type)) +geom_point() +scale_color_na() +scale_x_log10(name="Relative abundance of human virus reads") + theme_base +theme(axis.title.y =element_blank())g_ra_comp```# Human-infecting viruses: taxonomy and compositionI'm going to focus on the DNA reads here, since as discussed above I expect the RNA reads to be noisy and not very informative – which seems to in fact be the case.At the family level, as in Prussin, we see that *Papillomaviridae*, and *Polyomaviridae* dominate DNA reads in most samples, with *Poxviridae* and *Genomoviridae* showing significant presence in several samples. The first of these is primarily *Betapapillomavirus* and *Gammapapillomavirus*, with some *Alphapapillomavirus*; the second is primarily *Alphapolyomavirus* and *Deltapolyomavirus*. All of these are made up of several different viruses at the species level. *Genomoviridae* is represented primarily by *Gemykibivirus*, while *Poxviridae* is represented by small numbers of reads from several species, including vaccinia, cowpox and tanapox. (There's also one variola read, which would be alarming, but BLASTN thinks it isn't real.)Compared to Prussin, the most striking difference is the absence of herpesvirus reads; whereas HCMV was dominant in the Prussin data, it barely shows up here except in the controls.```{r}#| label: raise-hv-taxa# Get viral taxon names for putative HV readsviral_taxa$name[viral_taxa$taxid ==249588] <-"Mamastrovirus"viral_taxa$name[viral_taxa$taxid ==194960] <-"Kobuvirus"viral_taxa$name[viral_taxa$taxid ==688449] <-"Salivirus"viral_taxa$name[viral_taxa$taxid ==585893] <-"Picobirnaviridae"viral_taxa$name[viral_taxa$taxid ==333922] <-"Betapapillomavirus"viral_taxa$name[viral_taxa$taxid ==334207] <-"Betapapillomavirus 3"viral_taxa$name[viral_taxa$taxid ==369960] <-"Porcine type-C oncovirus"viral_taxa$name[viral_taxa$taxid ==333924] <-"Betapapillomavirus 2"viral_taxa$name[viral_taxa$taxid ==687329] <-"Anelloviridae"viral_taxa$name[viral_taxa$taxid ==325455] <-"Gammapapillomavirus"viral_taxa$name[viral_taxa$taxid ==333750] <-"Alphapapillomavirus"viral_taxa$name[viral_taxa$taxid ==694002] <-"Betacoronavirus"viral_taxa$name[viral_taxa$taxid ==334202] <-"Mupapillomavirus"viral_taxa$name[viral_taxa$taxid ==197911] <-"Alphainfluenzavirus"viral_taxa$name[viral_taxa$taxid ==186938] <-"Respirovirus"viral_taxa$name[viral_taxa$taxid ==333926] <-"Gammapapillomavirus 1"viral_taxa$name[viral_taxa$taxid ==337051] <-"Betapapillomavirus 1"viral_taxa$name[viral_taxa$taxid ==337043] <-"Alphapapillomavirus 4"viral_taxa$name[viral_taxa$taxid ==694003] <-"Betacoronavirus 1"viral_taxa$name[viral_taxa$taxid ==334204] <-"Mupapillomavirus 2"viral_taxa$name[viral_taxa$taxid ==334208] <-"Betapapillomavirus 4"viral_taxa$name[viral_taxa$taxid ==333928] <-"Gammapapillomavirus 2"viral_taxa$name[viral_taxa$taxid ==337039] <-"Alphapapillomavirus 2"mrg_hv_named <- mrg_hv %>%left_join(viral_taxa, by="taxid")# Discover viral species & genera for HV readsraise_rank <-function(read_db, taxid_db, out_rank ="species", verbose =FALSE){# Get higher ranks than search rank ranks <-c("subspecies", "species", "subgenus", "genus", "subfamily", "family", "suborder", "order", "class", "subphylum", "phylum", "kingdom", "superkingdom") rank_match <-which.max(ranks == out_rank) high_ranks <- ranks[rank_match:length(ranks)]# Merge read DB and taxid DB reads <- read_db %>%select(-parent_taxid, -rank, -name) %>%left_join(taxid_db, by="taxid")# Extract sequences that are already at appropriate rank reads_rank <-filter(reads, rank == out_rank)# Drop sequences at a higher rank and return unclassified sequences reads_norank <- reads %>%filter(rank != out_rank, !rank %in% high_ranks, !is.na(taxid))while(nrow(reads_norank) >0){ # As long as there are unclassified sequences...# Promote read taxids and re-merge with taxid DB, then re-classify and filter reads_remaining <- reads_norank %>%mutate(taxid = parent_taxid) %>%select(-parent_taxid, -rank, -name) %>%left_join(taxid_db, by="taxid") reads_rank <- reads_remaining %>%filter(rank == out_rank) %>%bind_rows(reads_rank) reads_norank <- reads_remaining %>%filter(rank != out_rank, !rank %in% high_ranks, !is.na(taxid)) }# Finally, extract and append reads that were excluded during the process reads_dropped <- reads %>%filter(!seq_id %in% reads_rank$seq_id) reads_out <- reads_rank %>%bind_rows(reads_dropped) %>%select(-parent_taxid, -rank, -name) %>%left_join(taxid_db, by="taxid")return(reads_out)}hv_reads_species <-raise_rank(mrg_hv_named, viral_taxa, "species")hv_reads_genus <-raise_rank(mrg_hv_named, viral_taxa, "genus")hv_reads_family <-raise_rank(mrg_hv_named, viral_taxa, "family")``````{r}#| label: hv-family#| fig-height: 6threshold_major_family <-0.06# Count reads for each human-viral familyhv_family_counts <- hv_reads_family %>%group_by(sample, room, sample_type, na_type, name, taxid) %>%count(name ="n_reads_hv") %>%group_by(sample, room, sample_type, na_type) %>%mutate(p_reads_hv = n_reads_hv/sum(n_reads_hv))# Identify high-ranking families and group othershv_family_major_tab <- hv_family_counts %>%group_by(name) %>%filter(p_reads_hv ==max(p_reads_hv)) %>%filter(row_number() ==1) %>%arrange(desc(p_reads_hv)) %>%filter(p_reads_hv > threshold_major_family)hv_family_counts_major <- hv_family_counts %>%mutate(name_display =ifelse(name %in% hv_family_major_tab$name, name, "Other")) %>%group_by(sample, room, sample_type, na_type, name_display) %>%summarize(n_reads_hv =sum(n_reads_hv), p_reads_hv =sum(p_reads_hv), .groups="drop") %>%mutate(name_display =factor(name_display, levels =c(hv_family_major_tab$name, "Other")))hv_family_counts_display <- hv_family_counts_major %>%rename(p_reads = p_reads_hv, classification = name_display)# Plotg_hv_family <- g_comp_base +geom_col(data=hv_family_counts_display, position ="stack") +scale_y_continuous(name="% HV Reads", limits=c(0,1.01), breaks =seq(0,1,0.2),expand=c(0,0), labels =function(y) y*100) +scale_fill_manual(values=palette_viral, name ="Viral family")g_hv_family``````{r}#| label: hv-genus#| fig-height: 6threshold_major_genus <-0.05# Count reads for each human-viral genushv_genus_counts <- hv_reads_genus %>%filter(na_type =="DNA") %>%group_by(sample, room, sample_type, na_type, name, taxid) %>%count(name ="n_reads_hv") %>%group_by(sample, room, sample_type, na_type) %>%mutate(p_reads_hv = n_reads_hv/sum(n_reads_hv))# Identify high-ranking families and group othershv_genus_major_tab <- hv_genus_counts %>%group_by(name) %>%filter(p_reads_hv ==max(p_reads_hv)) %>%filter(row_number() ==1) %>%arrange(desc(p_reads_hv)) %>%filter(p_reads_hv > threshold_major_genus)hv_genus_counts_major <- hv_genus_counts %>%mutate(name_display =ifelse(name %in% hv_genus_major_tab$name, name, "Other")) %>%group_by(sample, room, sample_type, na_type, name_display) %>%summarize(n_reads_hv =sum(n_reads_hv), p_reads_hv =sum(p_reads_hv), .groups="drop") %>%mutate(name_display =factor(name_display, levels =c(hv_genus_major_tab$name, "Other")))hv_genus_counts_display <- hv_genus_counts_major %>%rename(p_reads = p_reads_hv, classification = name_display)# Plotg_hv_genus <- g_comp_base +geom_col(data=hv_genus_counts_display, position ="stack") +scale_y_continuous(name="% HV Reads", limits=c(0,1.01), breaks =seq(0,1,0.2),expand=c(0,0), labels =function(y) y*100) +scale_fill_manual(values=palette_viral, name ="Viral genus")g_hv_genus``````{r}#| label: hv-species#| fig-height: 6threshold_major_species <-0.1# Count reads for each human-viral specieshv_species_counts <- hv_reads_species %>%filter(na_type =="DNA") %>%group_by(sample, room, sample_type, na_type, name, taxid) %>%count(name ="n_reads_hv") %>%group_by(sample, room, sample_type, na_type) %>%mutate(p_reads_hv = n_reads_hv/sum(n_reads_hv))# Identify high-ranking families and group othershv_species_major_tab <- hv_species_counts %>%group_by(name) %>%filter(p_reads_hv ==max(p_reads_hv)) %>%filter(row_number() ==1) %>%arrange(desc(p_reads_hv)) %>%filter(p_reads_hv > threshold_major_species)hv_species_counts_major <- hv_species_counts %>%mutate(name_display =ifelse(name %in% hv_species_major_tab$name, name, "Other")) %>%group_by(sample, room, sample_type, na_type, name_display) %>%summarize(n_reads_hv =sum(n_reads_hv), p_reads_hv =sum(p_reads_hv), .groups="drop") %>%mutate(name_display =factor(name_display, levels =c(hv_species_major_tab$name, "Other")))hv_species_counts_display <- hv_species_counts_major %>%rename(p_reads = p_reads_hv, classification = name_display)# Plotg_hv_species <- g_comp_base +geom_col(data=hv_species_counts_display, position ="stack") +scale_y_continuous(name="% HV Reads", limits=c(0,1.01), breaks =seq(0,1,0.2),expand=c(0,0), labels =function(y) y*100) +scale_fill_manual(values=palette_viral, name ="Viral species") +guides(fill=guide_legend(ncol=3))g_hv_species```Finally, here again are the overall relative abundances of the specific viral genera I picked out manually in my last entry:```{r}#| fig-height: 5#| label: ra-genera# Define reference generapath_genera_rna <-c("Mamastrovirus", "Enterovirus", "Salivirus", "Kobuvirus", "Norovirus", "Sapovirus", "Rotavirus", "Alphacoronavirus", "Betacoronavirus", "Alphainfluenzavirus", "Betainfluenzavirus", "Lentivirus")path_genera_dna <-c("Mastadenovirus", "Alphapolyomavirus", "Betapolyomavirus", "Alphapapillomavirus", "Betapapillomavirus", "Gammapapillomavirus", "Orthopoxvirus", "Simplexvirus","Lymphocryptovirus", "Cytomegalovirus", "Dependoparvovirus")path_genera <-bind_rows(tibble(name=path_genera_rna, genome_type="RNA genome"),tibble(name=path_genera_dna, genome_type="DNA genome")) %>%left_join(viral_taxa, by="name")# Count in each samplen_path_genera <- hv_reads_genus %>%group_by(sample, room, na_type, sample_type, name, taxid) %>%count(name="n_reads_viral") %>%inner_join(path_genera, by=c("name", "taxid")) %>%left_join(read_counts_raw, by=c("sample", "room", "na_type", "sample_type")) %>%mutate(p_reads_viral = n_reads_viral/n_reads_raw)# Pivot out and back to add zero linesn_path_genera_out <- n_path_genera %>% ungroup %>%select(sample, name, n_reads_viral) %>%pivot_wider(names_from="name", values_from="n_reads_viral", values_fill=0) %>%pivot_longer(-sample, names_to="name", values_to="n_reads_viral") %>%left_join(read_counts_raw, by="sample") %>%left_join(path_genera, by="name") %>%mutate(p_reads_viral = n_reads_viral/n_reads_raw)## Aggregate across datesn_path_genera_stype <- n_path_genera_out %>%group_by(na_type, sample_type, name, taxid, genome_type) %>%summarize(n_reads_raw =sum(n_reads_raw),n_reads_viral =sum(n_reads_viral), .groups ="drop") %>%mutate(sample="All samples", date="All dates",p_reads_viral = n_reads_viral/n_reads_raw)# Plotg_path_genera <-ggplot(n_path_genera_stype,aes(y=name, x=p_reads_viral, color=na_type)) +geom_point() +scale_x_log10(name="Relative abundance") +scale_color_na() +facet_grid(genome_type~sample_type, scales="free_y") + theme_base +theme(axis.title.y =element_blank())g_path_genera```One immediate observation, which I think helps demonstrate the unreliability of the RNA data here, is the almost complete absence of RNA-virus genera of interest. It's very suspicious for putative HIV reads to be almost as abundant as *Enterovirus* reads (which, remember, includes rhinovirus), while coronaviruses and influenza are totally absent. The DNA reads seem much more similar to Prussin, though I'm still surprised HCMV and *Mastadenovirus* are so rare.# ConclusionThis is the second, and by far the smaller, of the air-sampling datasets I've analyzed with this pipeline so far. Many of the high-level findings were similar to Prussin, including high relative abundance of human reads, low total viral reads, an absence of enteric viruses, and high abundance of papillomaviruses among human-infecting viruses. The biggest difference in the DNA data was the almost complete lack of cytomegalovirus, compared to its very high abundance in the Prussin data.Given how small this dataset is (16M reads on a MiSeq, compared to \~450M for Prussin and 1.3B for the upcoming [Leung et al.](https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-021-01044-7) dataset), I don't think too much weight should be placed on these findings, but given how rare air-MGS datasets are it's nice to have another one.