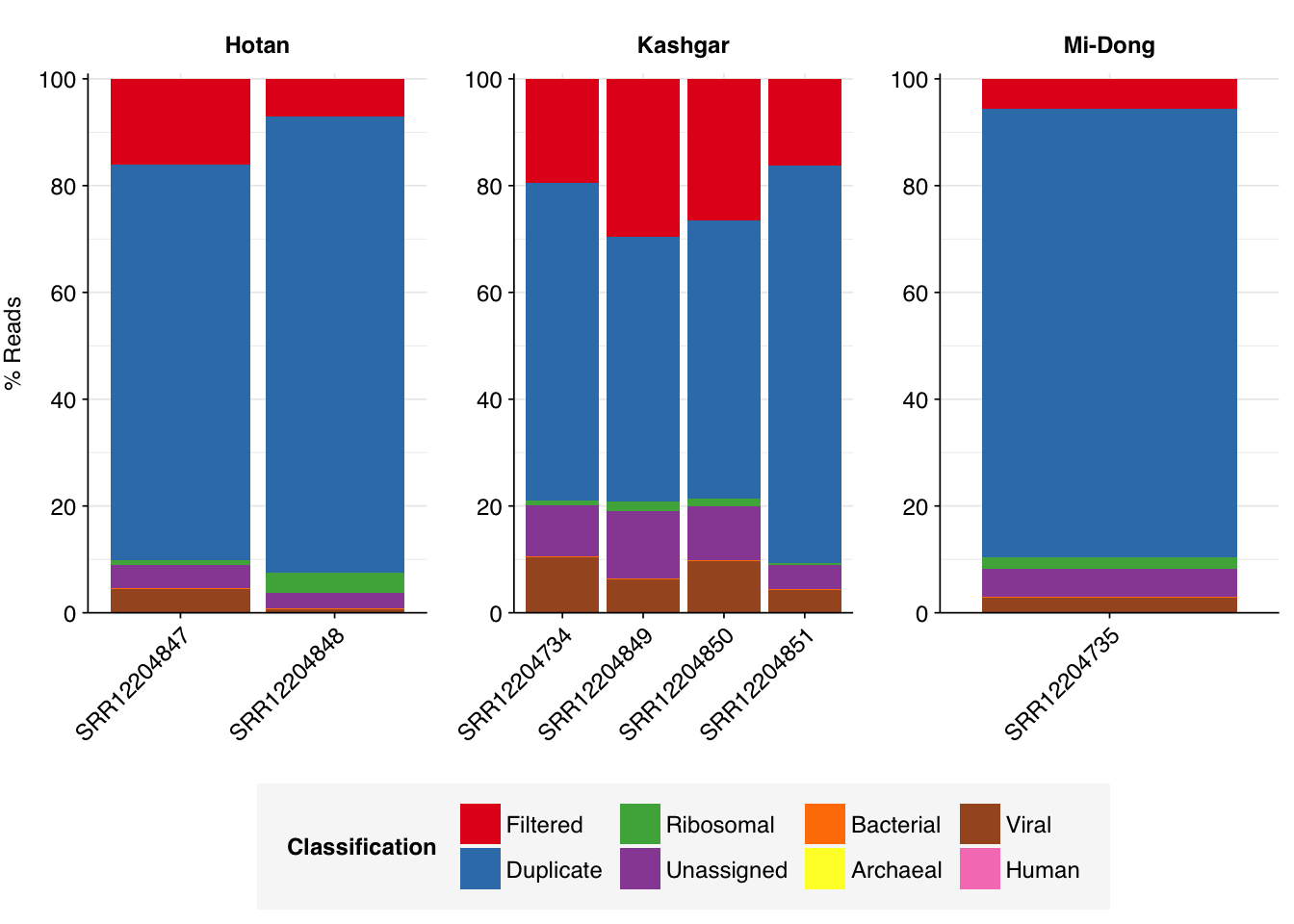
Some people had some helpful questions about my previous analysis of Yang et al. (2020), and the finding that the Yang data contains an unusually high relative abundance of viral sequences, together with an unusually low relative abundance of bacterial sequences. Here are the Bracken composition results I presented last time:
Multiple people noted that almost all of the reads in all samples were either removed during filtering or flagged as duplicates, and wondered whether the results would stand if we looked at the pre-deduplicated composition instead. Put another way, they wondered whether deduplication might be disproportionately removing ribosomal or other bacterial sequences, and thus giving a misleadingly optimistic picture of the results.
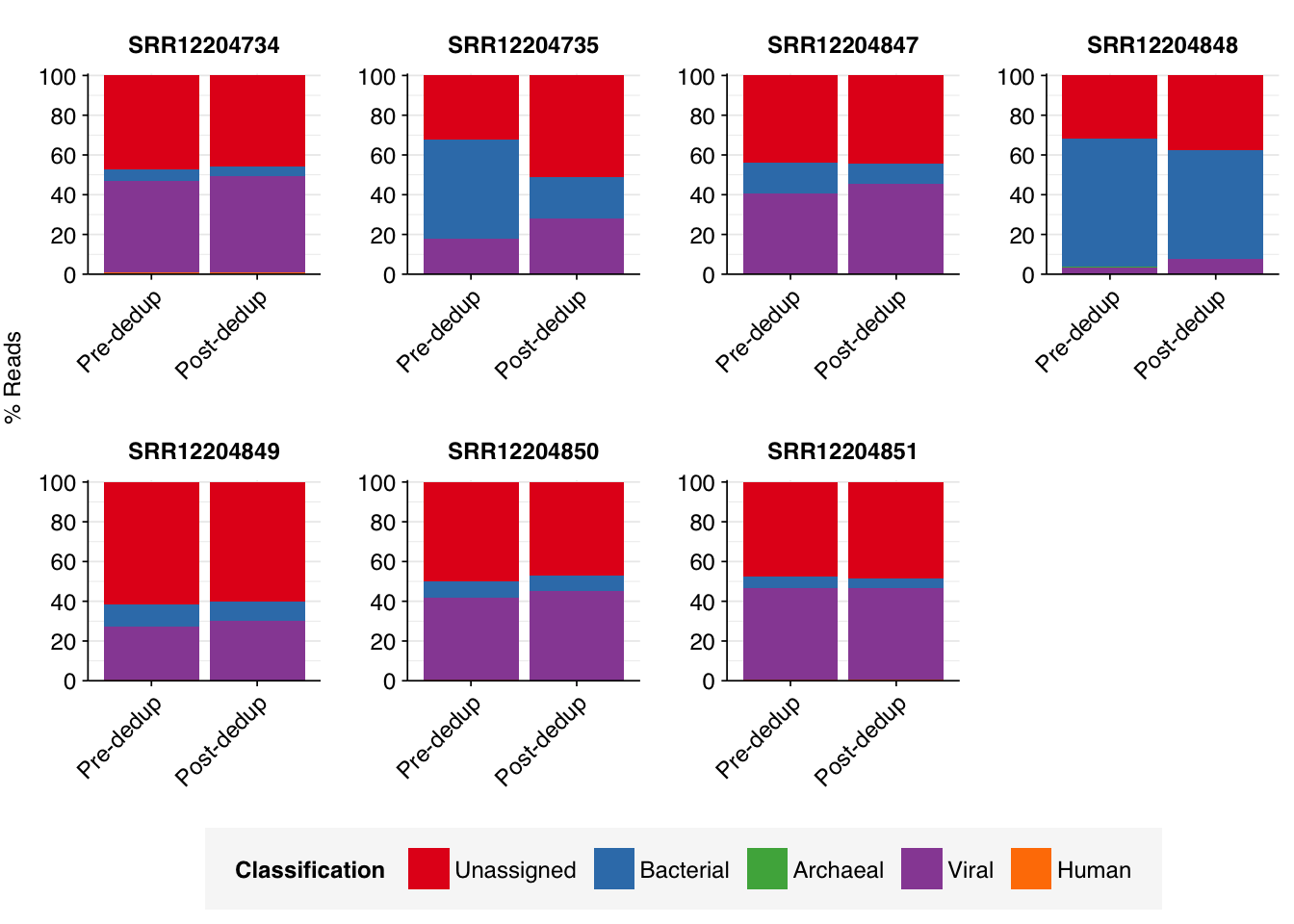
To address this, I re-ran the taxonomic composition analysis on a 1% subset of the pre-deduplication Yang data, then compared the effect of deduplication on the inferred taxonomic makeup (excluding filtered and deduplicated reads). In running this analysis, I made the assumption that all of the post-deduplication reads identified as ribosomal were from bacterial ribosomes; this seems to be generally true in our experience of wastewater data and allows us to make a more like-for-like comparison (as we don’t have ribosomal status for the pre-deduplication reads).
In most cases, deduplication made little-to-no difference to the inferred taxonomic composition, with bacterial and viral read fractions remaining roughly the same in both cases. In a few cases, and especially in sample SRR12204735, deduplication substantially reduced the inferred relative abundance of bacterial reads, probably due to collapsing genuine biological duplicates of common (e.g. ribosomal) sequences. In 5/7 samples, the viral fraction substantially exceeded the bacterial fraction both pre- and post-deduplication.
Overall, then, it appears that the finding of unusually high viral and low bacterial abundance in the Yang data is not an artefact of deduplication. This is a relief, and strengthens my confidence that their methods are worth investigating further.
Source Code
---title: "Followup analysis of Yang et al. (2020)"subtitle: "Digging into deduplication."author: "Will Bradshaw"date: 2024-03-19format: html: code-fold: true code-tools: true code-link: true df-print: pagededitor: visualtitle-block-banner: black---```{r}#| label: load-packages#| include: falselibrary(tidyverse)library(cowplot)library(patchwork)library(fastqcr)library(RColorBrewer)source("../scripts/aux_plot-theme.R")theme_base <- theme_base +theme(aspect.ratio =NULL)theme_kit <- theme_base +theme(axis.text.x =element_text(hjust =1, angle =45),axis.title.x =element_blank(),)tnl <-theme(legend.position ="none")```Some people had some helpful questions about my [previous analysis](https://data.securebio.org/wills-public-notebook/notebooks/2024-03-16_yang.html) of [Yang et al. (2020)](https://doi.org/10.1016/j.scitotenv.2020.142322), and the finding that the Yang data contains an unusually high relative abundance of viral sequences, together with an unusually low relative abundance of bacterial sequences. Here are the Bracken composition results I presented last time:```{r}data_dir <-"../data/2024-03-18_yang-2"data_dir_old <-file.path(data_dir, "yang-1")libraries_path <-file.path(data_dir_old, "sample-metadata.csv")basic_stats_path <-file.path(data_dir_old, "qc_basic_stats.tsv")stages <-c("raw_concat", "cleaned", "dedup", "ribo_initial", "ribo_secondary")libraries <-read_csv(libraries_path, show_col_types =FALSE)basic_stats <-read_tsv(basic_stats_path, show_col_types =FALSE) %>%inner_join(libraries, by="sample") %>%mutate(stage =factor(stage, levels = stages),sample =fct_inorder(sample))# Import Bracken databracken_path <-file.path(data_dir_old, "bracken_counts.tsv")bracken <-read_tsv(bracken_path, show_col_types =FALSE)total_assigned <- bracken %>%group_by(sample) %>%summarize(name ="Total",kraken_assigned_reads =sum(kraken_assigned_reads),added_reads =sum(added_reads),new_est_reads =sum(new_est_reads),fraction_total_reads =sum(fraction_total_reads))bracken_spread <- bracken %>%select(name, sample, new_est_reads) %>%mutate(name =tolower(name)) %>%pivot_wider(id_cols ="sample", names_from ="name", values_from ="new_est_reads")# Count readsread_counts_preproc <- basic_stats %>%select(sample, location, stage, n_read_pairs) %>%pivot_wider(id_cols =c("sample", "location"), names_from="stage", values_from="n_read_pairs")read_counts <- read_counts_preproc %>%inner_join(total_assigned %>%select(sample, new_est_reads), by ="sample") %>%rename(assigned = new_est_reads) %>%inner_join(bracken_spread, by="sample")# Assess compositionread_comp <-transmute(read_counts, sample=sample, location=location,n_filtered = raw_concat-cleaned,n_duplicate = cleaned-dedup,n_ribosomal = (dedup-ribo_initial) + (ribo_initial-ribo_secondary),n_unassigned = ribo_secondary-assigned,n_bacterial = bacteria,n_archaeal = archaea,n_viral = viruses,n_human = eukaryota)read_comp_long <-pivot_longer(read_comp, -(sample:location), names_to ="classification",names_prefix ="n_", values_to ="n_reads") %>%mutate(classification =fct_inorder(str_to_sentence(classification))) %>%group_by(sample) %>%mutate(p_reads = n_reads/sum(n_reads))read_comp_summ <- read_comp_long %>%group_by(location, classification) %>%summarize(n_reads =sum(n_reads), .groups ="drop_last") %>%mutate(n_reads =replace_na(n_reads,0),p_reads = n_reads/sum(n_reads),pc_reads = p_reads*100)# Plot overall compositiong_comp <-ggplot(read_comp_long, aes(x=sample, y=p_reads, fill=classification)) +geom_col(position ="stack") +scale_y_continuous(name ="% Reads", limits =c(0,1.01), breaks =seq(0,1,0.2),expand =c(0,0), labels =function(x) x*100) +scale_fill_brewer(palette ="Set1", name ="Classification") +facet_wrap(.~location, scales="free") + theme_kitg_comp```Multiple people noted that almost all of the reads in all samples were either removed during filtering or flagged as duplicates, and wondered whether the results would stand if we looked at the pre-deduplicated composition instead. Put another way, they wondered whether deduplication might be disproportionately removing ribosomal or other bacterial sequences, and thus giving a misleadingly optimistic picture of the results.To address this, I re-ran the taxonomic composition analysis on a 1% subset of the pre-deduplication Yang data, then compared the effect of deduplication on the inferred taxonomic makeup (excluding filtered and deduplicated reads). In running this analysis, I made the assumption that all of the post-deduplication reads identified as ribosomal were from bacterial ribosomes; this seems to be generally true in our experience of wastewater data and allows us to make a more like-for-like comparison (as we don't have ribosomal status for the pre-deduplication reads).The results were as follows:```{r}class_levels <-c("Unassigned", "Bacterial", "Archaeal", "Viral", "Human")# 1. Remove filtered & duplicate reads from original Bracken output & renormalizeread_comp_renorm <- read_comp_long %>%filter(! classification %in%c("Filtered", "Duplicate")) %>%group_by(sample) %>%mutate(p_reads = n_reads/sum(n_reads),classification = classification %>% as.character %>%ifelse(. =="Ribosomal", "Bacterial", .)) %>%group_by(sample, location, classification) %>%summarize(n_reads =sum(n_reads), p_reads =sum(p_reads), .groups ="drop") %>%mutate(classification =factor(classification, levels = class_levels))# 2. Import pre-deduplicated bracken_path_predup <-file.path(data_dir, "bracken_counts_subset.tsv")bracken_predup <-read_tsv(bracken_path_predup, show_col_types =FALSE)total_assigned_predup <- bracken_predup %>%group_by(sample) %>%summarize(name ="Total",kraken_assigned_reads =sum(kraken_assigned_reads),added_reads =sum(added_reads),new_est_reads =sum(new_est_reads),fraction_total_reads =sum(fraction_total_reads))bracken_spread_predup <- bracken_predup %>%select(name, sample, new_est_reads) %>%mutate(name =tolower(name)) %>%pivot_wider(id_cols ="sample", names_from ="name", values_from ="new_est_reads")# Count readsread_counts_predup <- read_counts_preproc %>%select(sample, location, raw_concat, cleaned) %>%mutate(raw_concat = raw_concat *0.01, cleaned = cleaned *0.01) %>%inner_join(total_assigned_predup %>%select(sample, new_est_reads), by ="sample") %>%rename(assigned = new_est_reads) %>%inner_join(bracken_spread_predup, by="sample")# Assess compositionread_comp_predup <-transmute(read_counts_predup, sample=sample, location=location,n_filtered = raw_concat-cleaned,n_unassigned = cleaned-assigned,n_bacterial = bacteria,n_archaeal = archaea,n_viral = viruses,n_human = eukaryota)read_comp_predup_long <-pivot_longer(read_comp_predup, -(sample:location), names_to ="classification",names_prefix ="n_", values_to ="n_reads") %>%mutate(classification =fct_inorder(str_to_sentence(classification))) %>%group_by(sample) %>%mutate(p_reads = n_reads/sum(n_reads)) %>%filter(! classification %in%c("Filtered", "Duplicate")) %>%group_by(sample) %>%mutate(p_reads = n_reads/sum(n_reads))# 3. Combineread_comp_comb <-bind_rows(read_comp_predup_long %>%mutate(deduplicated =FALSE), read_comp_renorm %>%mutate(deduplicated =TRUE)) %>%mutate(label =ifelse(deduplicated, "Post-dedup", "Pre-dedup") %>% fct_inorder)# Plot overall compositiong_comp_predup <-ggplot(read_comp_comb, aes(x=label, y=p_reads, fill=classification)) +geom_col(position ="stack") +scale_y_continuous(name ="% Reads", limits =c(0,1.01), breaks =seq(0,1,0.2),expand =c(0,0), labels =function(x) x*100) +scale_fill_brewer(palette ="Set1", name ="Classification") +facet_wrap(.~sample, scales="free", ncol=4) + theme_kitg_comp_predup```In most cases, deduplication made little-to-no difference to the inferred taxonomic composition, with bacterial and viral read fractions remaining roughly the same in both cases. In a few cases, and especially in sample SRR12204735, deduplication substantially reduced the inferred relative abundance of bacterial reads, probably due to collapsing genuine biological duplicates of common (e.g. ribosomal) sequences. In 5/7 samples, the viral fraction substantially exceeded the bacterial fraction both pre- and post-deduplication.Overall, then, it appears that the finding of unusually high viral and low bacterial abundance in the Yang data is not an artefact of deduplication. This is a relief, and strengthens my confidence that their methods are worth investigating further.