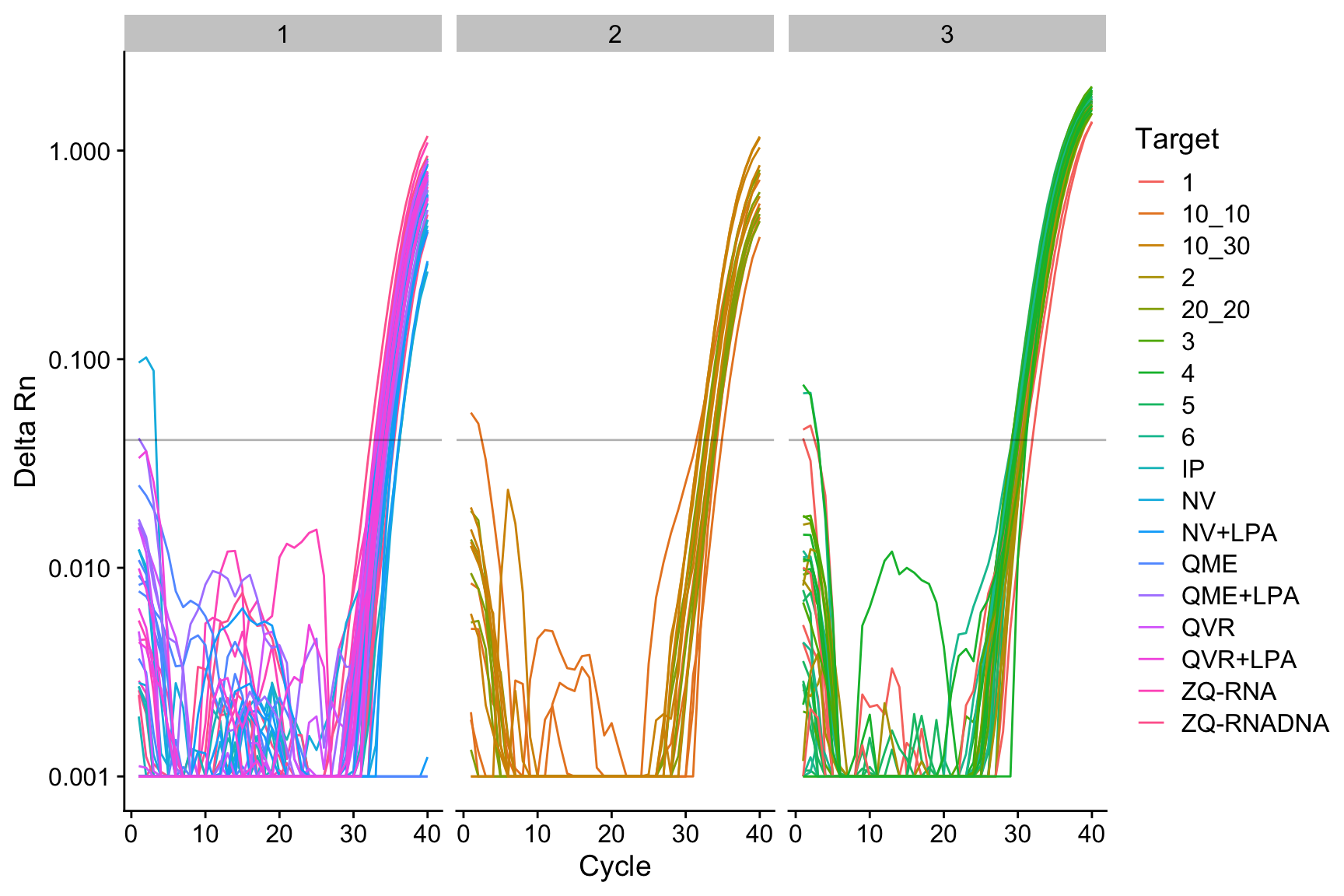
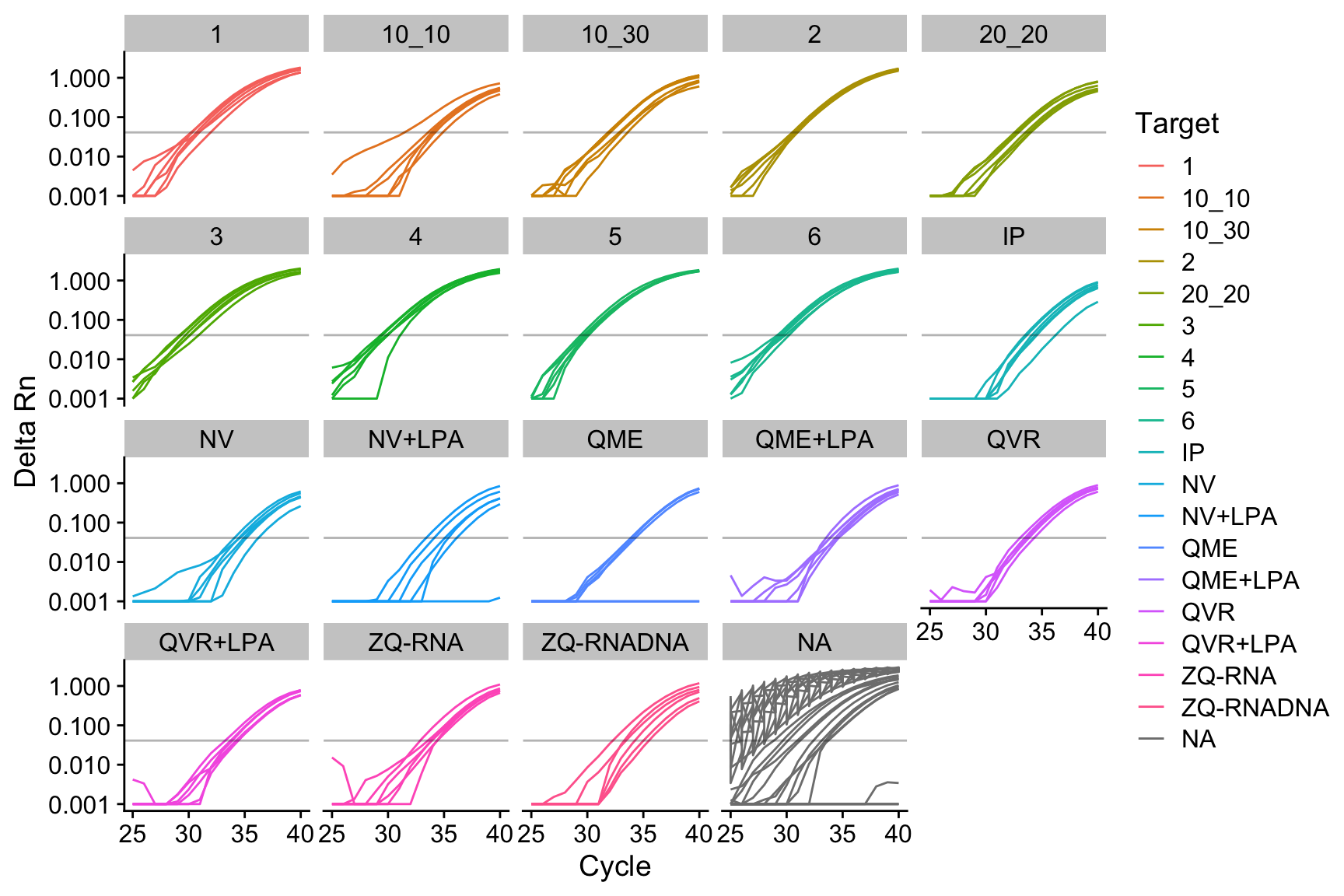
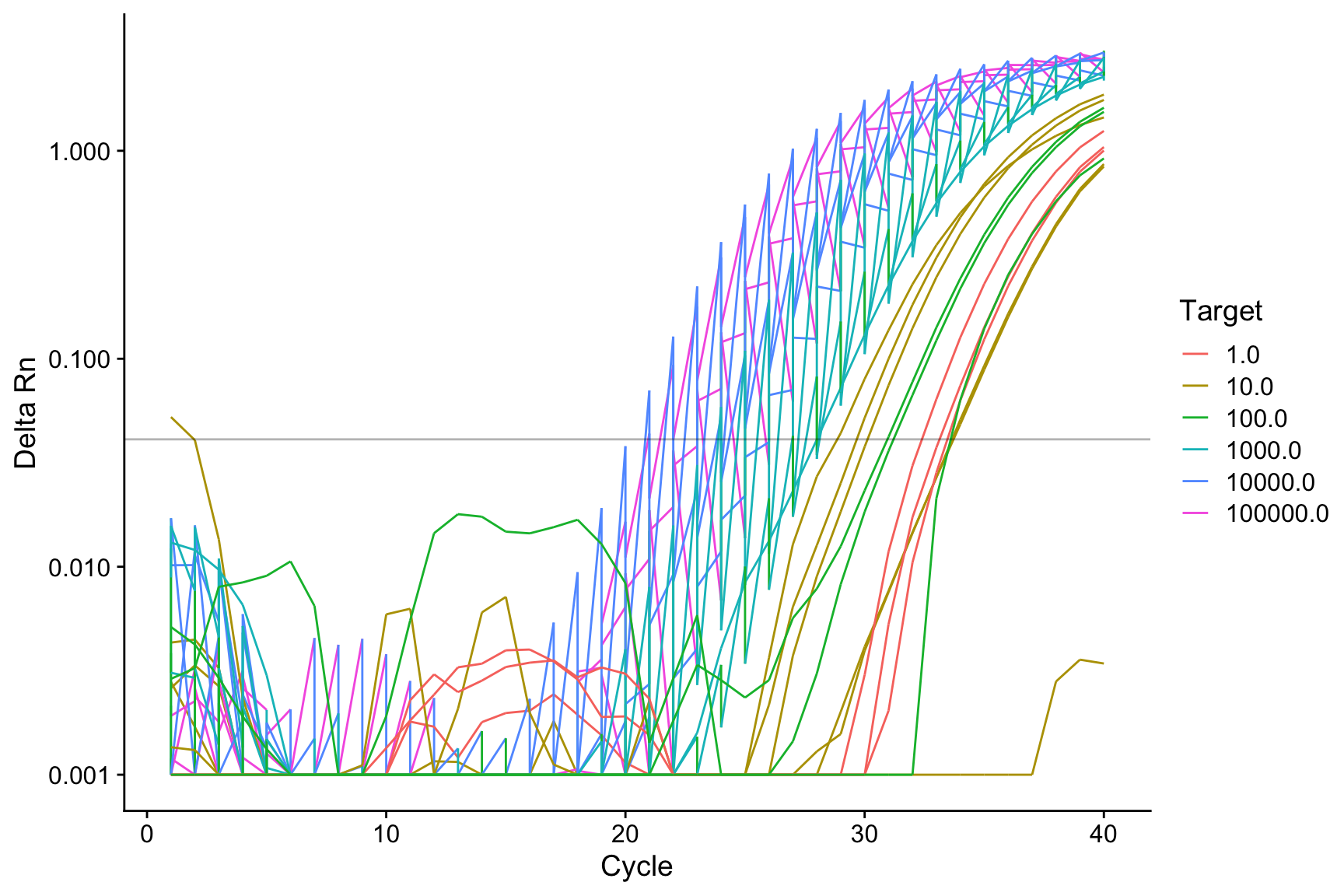
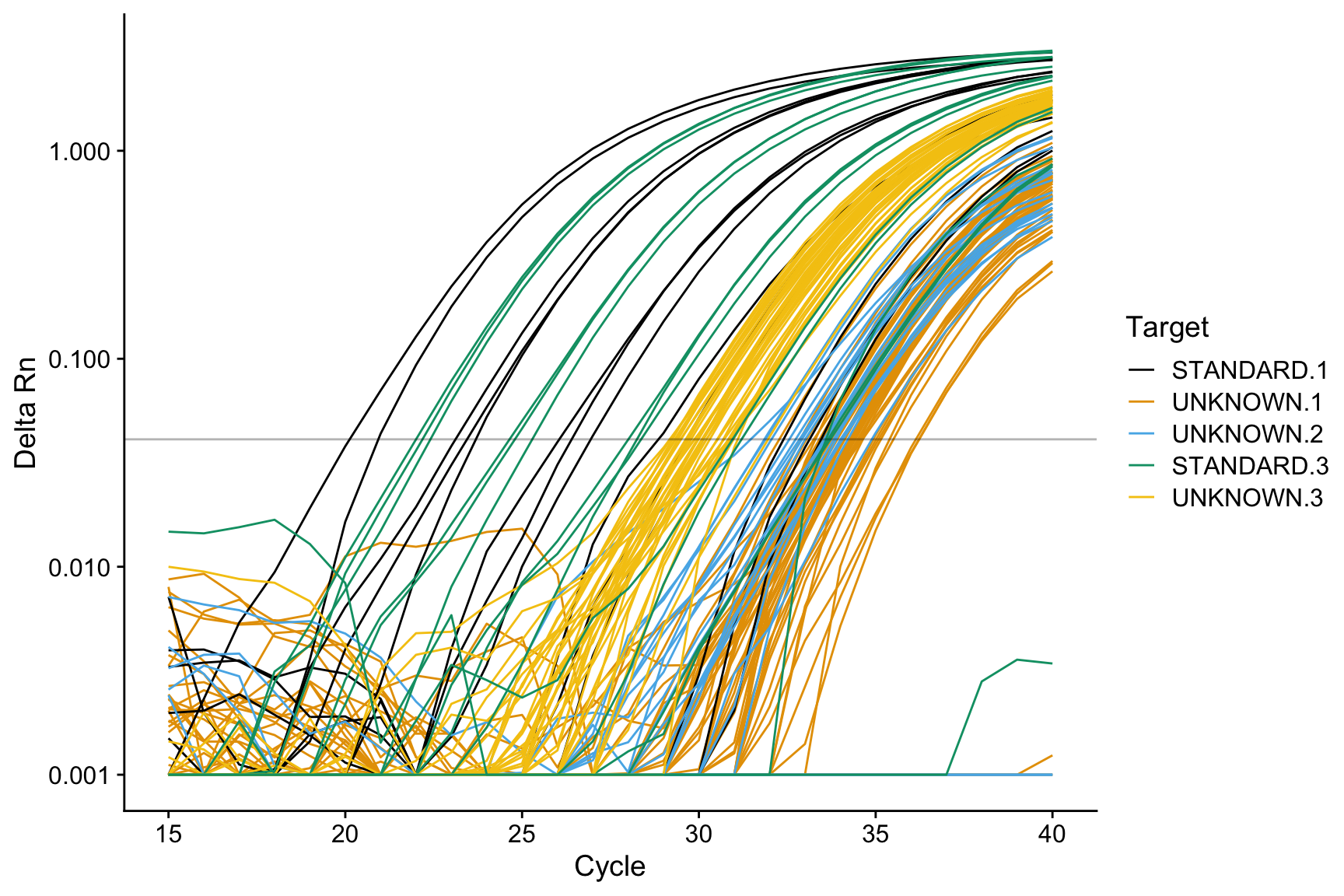
Rows: 27 Columns: 8
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (7): sample_qPCR, Kit, treatment_group, LPA, other_treatment, Virus_spec...
dbl (1): group
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Rows: 27
Columns: 8
$ sample_qpcr <chr> "1_ZR_A", "1_ZR_B", "1_ZR_C", "2_ZDR_A", "2_ZDR_B", "2…
$ kit <chr> "Zymo RNA", "Zymo RNA", "Zymo RNA", "Zymo RNA/DNA", "Z…
$ treatment_group <chr> "ZQ-RNA", "ZQ-RNA", "ZQ-RNA", "ZQ-RNADNA", "ZQ-RNADNA"…
$ lpa <chr> "No", "No", "No", "No", "No", "No", "No", "No", "No", …
$ group <dbl> 3, 3, 3, 3, 3, 3, 3, 3, 3, 1, 1, 1, 1, 1, 1, 1, 1, 1, …
$ other_treatment <chr> "Shield", "Shield", "Shield", "Shield", "Shield", "Shi…
$ virus_specific <chr> "Yes", "Yes", "Yes", "Yes", "Yes", "Yes", "No", "No", …
$ fp_data <chr> "No", "Yes", "No", "No", "Yes", "No", "No", "Yes", "No…
Rows: 5
Columns: 2
$ target_qpcr <chr> "CrA", "Cov2", "Noro", "16S", "Phg"
$ target <chr> "Crassphage", "SARS-CoV-2", "Norovirus", "bacteria", "phag…